一种降解木质素的锰过氧化物酶基因及其获取方法与流程
本发明涉及分子生物
技术领域:
,尤其涉及一种降解木质素的锰过氧化物酶基因及其获取方法。
背景技术:
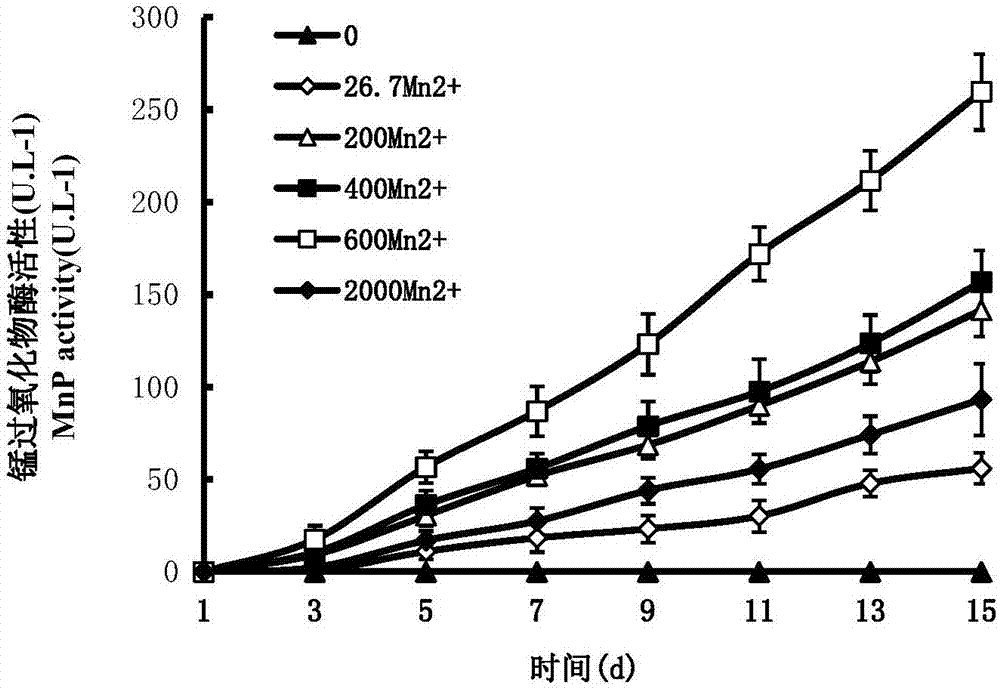
:近年来二次污染问题愈发严峻,主要表现为一些特殊异生物质和排放污染物难以被生物降解,不少国家正在积极寻求省资源、少污染的新技术。当前生物制浆造纸的一大难题是脱除木材中的木质素,使木材成浆中纤维材料的氢键相结合来实现纸张的有效粘结。利用白腐菌中的木质素降解酶对木质素进行生物降解,最终能把木质素彻底降解为CO2和H2O,就从根本上解决了造纸工业的环境污染问题。在木材上生长的树舌,就是我们常见的白色腐朽菌,它不仅寄生在腐朽木材上,而且还寄生在活立木上,可见白腐菌树舌具有降解木质素的潜在能力。木材腐朽菌在分解木质素的过程中,能够产生一系列胞外氧化酶,其中最主要的有锰过氧化物酶((Manganeseperoxida简称MnP)、木质素过氧化物酶(Ligninperoxidas简称LiP)、和漆酶(Laccase)。锰过氧化物酶(MnP)是一种依赖H2O2的亚铁血红素糖蛋白酶,在真菌降解木质素的非特异性胞外氧化还原酶中,MnP起着至关重要的作用。它可以降解木质素及苯酚类物质,还可攻击各种有机污染物包括持久稳固的化学农药、染料脱色等。在世界范围内,木材腐朽菌易导致多种木材发生病害,给林业生产造成了严重损失,在此基础上,以重要木质素降解酶-MnP为研究对象,借助酶活和基因的操作技术,探明锰过氧化物酶基因的功能特性,创建其研究技术体系,对预期的研究具重要的理论与实践意义。因此,研究树舌Ga-MnP基因及其酶活的特性,以便提高MnP的产量与降解活力成为一个急需解决的重要问题。随着分子生物技术的不断进步,科研工作者已开始研究MnP的应用。目前该方面报道不多,锰过氧化物酶系统的检测工作及其基因的研究也鲜见报道。技术实现要素:本发明的目的在于克服现有技术的不足,提供了一种降解木质素的锰过氧化物酶基因及其获取方法,特别涉及该基因的PCR扩增,逆转录RT-PCR及cDNA末端的快速扩增(RACE)技术等方法,扩增出Ga-MnP全长cDNA序列,证明了该基因与其它白腐菌锰过氧化物酶基因具有96%的同源性,MnP基因可引起木材的腐朽,对木质素具有降解作用,由此证明了Ga-MnP基因是锰过氧化物酶基因家簇的一个重要成员,本发明为进一步研制生物催化剂提供技术储备,以达到工业高效生产的需求。本发明是通过以下技术方案实现的:一种降解木质素的锰过氧化物酶基因,来源于白腐菌树舌菌株(G.applanatum)AQ1,所述白腐菌树舌菌株在中国典型培养物保藏中心(CCTCC)的保藏编号为CCTCCM2017017;所述降解木质素的锰过氧化物酶基因的基因序列如SeqIDNo:1所示,蛋白质氨基酸序列如SeqIDNO:2所示。一种降解木质素的锰过氧化物酶基因的获取方法,包括以下步骤:(1)利用白腐菌树舌菌株,菌种培养至10-15d,将培养的白腐菌树舌菌丝接种于LNAS液体培养基培养中,再于A、B、C、D四种不同培养条件下对其进行进一步地诱导培养;其中,不含Mn2+、不加底物的培养基条件为A,含Mn2+、不加底物的培养基条件为B,含Mn2+的LNAS填加底物木屑的培养基条件为C,含Mn2+填加底物2,6-DMP的培养基条件为D;接着,于0-21天内分别在同一时间收获菌液,将菌液离心沉淀后,再以2,6-DMP为底物检测胞内锰过氧化物酶酶活力;最后,根据四种培养条件下锰过氧化物酶酶活力的比较,选择出酶活力最高、产酶量最大的C培养条件作为最佳培养条件;(2)培养白腐菌树舌菌株,菌种培养至10-15d时,将培养的白腐菌树舌菌丝接种于LNAS液体培养基培养中,采取C培养条件进行诱导培养,且在诱导培养过程中分别添加不同浓度的Mn2+进行诱导培养,再于0-21天内分别在同一时间收获菌液,将菌液离心沉淀后,再以2,6-DMP为底物,检测不同浓度的Mn2+下胞内锰过氧化物酶酶活力;(3)提取白腐菌树舌基因组总DNA,对比已报道的其它白腐菌锰过氧化物酶同源基因序列,进行相似序列比对分析,界定该类基因的保守序列LTFHDAI和HTVGSQN,再设计简并引物Ga-MnPF、Ga-MnPR,通过简并PCR反应获得Ga-MnP目的基因片段;(4)提取白腐菌树舌总RNA,通过反转录合成cDNA第一链,再将目的基因PCR产物凝胶回收并与T载体连接,将连接后的产物进行转化,再进行菌落PCR和质粒PCR平行验证;(5)利用RACE技术,扩增出Ga-MnP特定位点到3’或到5’端之间的未知核苷酸序列,从cDNA中分离目的基因Ga-MnP,阳性克隆测序获得基因全长序列;(6)利用逆转录RT-PCR技术对该基因Ga-MnP进行扩增,并将所扩增的基因片段连接于pMD20-T载体,进行基因序列的测定;(7)将所得核酸序列翻译成氨基酸序列,与相近的白腐菌木质素降解酶和锰过氧化物酶基因的同源序列进行对比,对其进行生物信息学的分析。作为本发明的优选方式之一,所述白腐菌树舌菌株源于杭州。作为本发明的优选方式之一,所述步骤(1)和步骤(2)中LNAS液体培养基的组成配方为(1000mL):0.2gKH2PO4,0.5gMgSO4·7H2O,1.18g琥珀酸,0.1gCaCl2·2H2O,1ml矿质溶液,19.4mL氮溶液,0.5mL维他命溶液,pH4.5;矿质溶液为两种,一种为含Mn2+矿质溶液,一种为不含Mn2+矿质溶液;当采用B、C或D培养条件进行诱导培养时,前期菌种培养采用的LNAS液体培养基中的矿质溶液为含Mn2+矿质溶液,其组成配方为(1000mL):0.087g柠檬酸铁,0.1gZnSO4·7H2O,0.45gMnSO4·H2O,0.1gCoCl2·6H2O,0.01gCuSO4·5H2O;当采取A培养条件进行诱导培养时,前期菌种培养采用的LNAS液体培养基中的矿质溶液为不含Mn2+矿质溶液,其组成配方为(1000mL):0.087g柠檬酸铁,0.1gZnSO4·7H2O,0.1gCoCl2·6H2O,0.01gCuSO4·5H2O;氮溶液的组成配方为(1000mL):4.0gL-天门冬素,2.0gNH4NO3;维他命溶液的组成配方为(1000mL):0.002gvitaminH,0.002gvitaminM,0.005gvitaminB1,0.005gvitaminB2,0.01gvitaminB6,0.0001gvitaminB12,0.005g烟酸,0.005g泛酸钙,0.005g对氨基苯甲酸,0.005gDL-6,8-硫辛酸。作为本发明的优选方式之一,所述步骤(1)中木屑为青杨木屑;所述步骤(1)和步骤(2)中的Mn2+具体为硫酸锰溶液,所述步骤(2)中诱导培养过程中添加的硫酸锰溶液浓度分别为0、26.7、200、400、600和2000μmol·L-1。作为本发明的优选方式之一,所述步骤(1)和步骤(2)中以2,6-DMP为底物检测锰过氧化物酶酶活力的具体方法为:先配制酶活反应体系(1mL):50μL10mmol·L-1的反应底物2,6-DMP(2,6—二甲氧基苯酚),840μL50mmol·L-1丙二酸钠缓冲液,50μL10mmol·L-1MnSO4,50μL酶液样品和10μL10mmol·L-1H2O2,启动反应,再于470nm处测定吸光度在1min内的反应液吸光值A470。作为本发明的优选方式之一,所述步骤(3)中提取白腐菌树舌基因组总DNA的具体步骤为:(1)取新鲜白腐菌树舌菌丝15-25mg,放入研钵中加入液氮充分碾磨,加入400μl缓冲液FP1和6μl的RNaseA(10mg/mL),旋涡振荡0.5-1.5min,室温放置5-15min;(2)加入130μl缓冲液FP2,充分混匀,涡旋振荡0.5-1.5min,11000-13000rpm离心4-6min,将上清转移至新的离心管中;(3)向上清液中加入0.7倍体积的异丙醇,充分混匀,此时出现絮状基因组DNA,11000-13000rpm离心1-3min,弃上清,保留沉淀;(4)加入700μl70%乙醇,涡旋振荡4-6s,11000-13000rpm离心1-3min,弃上清;(5)重复步骤4;(6)开盖倒置,室温5-10min,彻底晾干残余的乙醇,加入适量洗脱缓冲液TE,65℃水浴10-60min溶解DNA,期间颠倒混匀助溶,最终得到DNA溶液;(7)采用1%琼脂糖凝胶电泳检测完整性,检测后的基因组DNA置于-20℃冰箱保存。作为本发明的优选方式之一,所述步骤(3)中利用软件ClustalX2.0进行相似序列比对分析;所述步骤(7)中利用NCBI中BLASTP软件将核酸序列翻译成氨基酸序列。作为本发明的优选方式之一,所述步骤(3)中Ga-MnPF引物的序列为:5'-CTGACCTTCCATGACGCTAT-3';Ga-MnPR引物的序列为:5'-GTATGGCAGCCAAGCGTAGGG-3'。作为本发明的优选方式之一,所述步骤(4)中提取白腐菌树舌基因组总RNA的具体步骤为:(1)取小于100mg的白腐菌树舌菌丝放入研钵中,加液氮研磨成糊状,用小药匙刮研钵壁上研磨好的组织并转移到离心管内;(2)将研磨好的白腐菌树舌菌丝放入离心管后,立即添加600μlBufferRB/β-巯基乙醇;(3)用枪头将裂解液迅速移入到放置在2mL离心管的Homogenizationspincolumn中,室温12000-14000×g离心4-6min;(4)转移上清液到一个新的离心管内,加入0.5倍体积的无水乙醇;(5)取一个HiBindRNA小量柱,并把柱子装在一个收集管内,转移混合液至柱子中,轻轻的盖上管帽,室温下9000-11000×g离心25-35s,弃去收集管中的滤液,并把柱子重新装在收集管中;(6)把柱子重新放入新的收集管中,添加300μlRNA洗涤Buffer1到柱中,室温9000-11000×g离心0.5-1.5min;(7)将75μlDNase1消化处理混合剂立即准确加入到每个HiBidRNA柱膜的中央,室温下(25-30℃)放置14-16min;(8)将柱子放置在新的收集管上,添加500μlRNA洗涤Buffer1,室温静置4-6min,室温下,9000-11000×g离心0.5-1.5min,弃滤液,重新使用一个收集管;(9)将柱子放置在同样的收集管上,添加500μlRNA洗涤Buffer2,室温下,9000-11000×g离心0.5-1.5min,弃滤液,再次使用收集管;(10)用500μlRNA洗涤Buffer2第二次洗涤柱子,重复上一步骤,室温下,9000-11000×g离心0.5-1.5min,弃滤液,然后使用新的收集管,室温下,9000-11000×g离心空柱0.5-1.5min,以甩干柱子的基质;(11)洗脱RNA,将柱子转移到新的离心管上并且用50-100μl的DEPC水洗涤RNA,将提取的白腐菌树舌总RNA溶液贮存于-80℃保存备用。本发明相比现有技术的优点在于:本发明研究锰过氧化物酶的酶活检测方法,获得最佳的培养基成分和酶作用的底物关系,检测出在单位体积下,降解木质素的锰过氧化物酶最大酶活性;此外,在检测木质素降解酶系统的主要酶系和酶学性质的前提下,产生一个同源性为96%的锰过氧化物酶基因,通过简并/特异性引物的准确定位,不断的PCR扩增及验证试验,克隆出了Ga-MnP基因全长cDNA序列。附图说明图1是实施例2中的A、B、C、D四种不同培养条件下白腐菌树舌胞外锰过氧化物酶酶活的比较图;图2是实施例1中不同浓度的Mn2+下胞内锰过氧化物酶酶活力比较图;图3为实施例3中提取的白腐菌树舌基因组的电泳检测图(泳道1,2为基因组总DNA,泳道M为Maker);图4为实施例4中利用简并PCR方法扩增筛选的目的基因Ga-MnP基因片段的电泳检测图(泳道1,2为目的基因Ga-MnP基因片段,泳道M为Maker);图5为实施例5中提取的白腐菌树舌总RNA的电泳检测图(泳道1,2为基因组总RNA,泳道M为Maker);图6为实施例6中特异性引物进行反转录扩增出的Ga-MnP基因cDNA片段的电泳检测图(泳道2,3为扩增出的Ga-MnP基因cDNA片段,泳道M为Maker);图7为实施例9中以反转录的cDNA为模板用3′RACE特异性引物进行的套式PCR扩增的PCR产物的电泳检测图(泳道2,3为PCR产物,泳道M为Maker);图8为实施例9以反转录的cDNA为模板用5′RACE特异性引物进行的PCR扩增的PCR产物的电泳检测图(泳道2,3为PCR产物,泳道M为Maker);图9为实施例8以反转录的cDNA为模板,将5′和3′RACE拼接的序列,进行Ga-MnP基因全长cDNA序列的扩增验证,PCR产物经1%的琼脂糖凝胶电泳检测(泳道2,3为PCR产物,泳道M为Maker)。具体实施方式下面对本发明的实施例作详细说明,本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。实施例1本实施例说明一种降解木质素的锰过氧化物酶基因:一种降解木质素的锰过氧化物酶基因,来源于白腐菌树舌菌株(Ganodermaapplanatum)AQ1,该白腐菌树舌菌株在中国典型培养物保藏中心(CCTCC)的保藏编号为CCTCCM2017017;该降解木质素的锰过氧化物酶基因的基因序列如SeqIDNo:1所示,蛋白质氨基酸序列如SeqIDNO:2所示;该基因编码的Ga-MnP具有降解木质素的功能可导致木材腐朽变质。该基因Ga-MnP的cDNA全长1301bps,完整开放阅读框含1095个碱基,编码一种含有364个氨基酸的蛋白质,起始密码子ATG,终止密码子TGA,5′端非翻译区有58个核苷酸,3′端非翻译区有148个核苷酸,分子量为38.35kDa,GC含量为61.92,等电点为4.23,Ga-MnP蛋白质多肽前体包含1个18氨基酸的信号肽及1个8氨基酸的中间前导短肽。N末端序列为M(Met),蛋白不稳定指数为43.69,脂肪族指数为43.69,亲水性为0.084,此蛋白为亲水蛋白。实施例2本实施例说明来源于白腐菌树舌菌株的锰过氧化物酶酶活检测方法:(1)利用白腐菌树舌菌株(源于杭州分离后保藏),菌种培养至13d,将培养的白腐菌树舌菌丝接种于LNAS液体培养基培养中,再于A、B、C、D四种不同培养条件下对其进行进一步地诱导培养;其中,不含Mn2+(硫酸锰)、不加底物的培养基条件为A,含Mn2+(硫酸锰)、不加底物的培养基条件为B,含Mn2+(硫酸锰)的LNAS填加底物青杨木屑的培养基条件为C,含Mn2+(硫酸锰)添加底物2,6-DMP的培养基条件为D;接着,于0-21天内分别在同一时间收获菌液,将菌液离心沉淀后,再以2,6-DMP为底物检测胞内锰过氧化物酶酶活力(先配制酶活反应体系1mL:50μL10mmol·L-1的反应底物2,6-DMP,840μL50mmol·L-1丙二酸钠缓冲液,50μL10mmol·L-1MnSO4,50μL酶液样品和10μL10mmol·L-1H2O2,启动反应,再于470nm处测定吸光度在1min内的反应液吸光值A470);图1为A、B、C、D四种不同培养条件下白腐菌树舌胞外锰过氧化物酶酶活的比较图,图1表明:在培养方式A中,提取的酶液未检测到MnP的活性;在培养方式B中,提取的酶液可以检测到MnP的活性变化,但活性很低;在培养方式C中,提取的酶液检测到MnP的活性变化,MnP在13天达到最大分泌量,酶活力为45.56U·L-1;在培养方式D中,提取的酶液也检测到MnP的活性变化,MnP在15天达到最大分泌量,酶活力为4.84U·L-1;对比C,D两种培养过程中MnP产生结果,表明在培养方式C和D中分别添加2种底物木屑和2,6-DMP可以提高MnP的产生量,底物对于该菌木质素降解酶系统可产生诱导作用。以木屑为底物MnP产量要高于以2,6-DMP为底物的产生量,其吸收峰在470nm处有明显的峰值变化,而其他3种培养方式的吸光度和吸收峰表现为不明显的变化;最后,根据四种培养条件下锰过氧化物酶酶活力的比较,可选择出酶活力最高、产酶量最大的C培养条件作为最佳培养条件用于后续实验;(2)培养白腐菌树舌菌株,菌种培养至13d时,将培养的白腐菌树舌菌丝接种于LNAS液体培养基培养中,采取C培养条件进行诱导培养,且在诱导培养过程中分别添加不同浓度的Mn2+(0、26.7、200、400、600和2000μmol·L-1的硫酸锰溶液)进行诱导培养,再于0-21天内分别在同一时间收获菌液;将菌液离心沉淀后,再以2,6-DMP为底物,检测不同浓度的Mn2+下胞内锰过氧化物酶酶活力(先配制酶活反应体系1mL:50μL10mmol·L-1的反应底物2,6-DMP,840μL50mmol·L-1丙二酸钠缓冲液,50μL10mmol·L-1MnSO4,50μL酶液样品和10μL10mmol·L-1H2O2,启动反应,再于470nm处测定吸光度在1min内的反应液吸光值A470);图2为不同浓度的Mn2+下胞内锰过氧化物酶酶活力比较图,图2表明:在Mn2+的浓度为26.7μmol·L-1,提取的酶液中MnP的活性在15d达到最大分泌量,酶活力为55.92U·L-1;在Mn2+的浓度为200μmol·L-1,提取的酶液中MnP的活性在15d达到最大分泌量,酶活力为141.51U·L-1;在Mn2+的浓度为400μmol·L-1,提取的酶液中MnP的活性在15d达到最大分泌量,酶活力为156.71U·L-1;在Mn2+的浓度为600μmol·L-1,提取的酶液中MnP的活性在15d达到最大分泌量,酶活力为259.55U·L-1;在Mn2+的浓度为2000μmol·L-1,提取的酶液中MnP的活性在15d达到最大分泌量,酶活力为93.14U·L-1;结果表明不同的Mn2+的培养方式变化是有规律的:不含Mn2+的培养方式下,21d均未检测到MnP的活性变化,而Mn2+的浓度分别为(26.7、200、400和600μmol·L-1),MnP的活性在3d之后产生,随之逐渐升高,在第15d达到最高,而Mn2+的浓度为2000μmol·L-1时,MnP的活性没有Mn2+的浓度400和600μmol·L-1的活性高,表明Mn2+的浓度达到一定程度时,MnP的活性会下降;不同的Mn2+的培养方式对比,可见Mn2+是白腐菌产生MnP的必要因子,在诱导MnP的产生过程中起着关键作用。进一步地,LNAS液体培养基的组成配方为(1000mL):0.2gKH2PO4,0.5gMgSO4·7H2O,1.18g琥珀酸,0.1gCaCl2·2H2O,1ml矿质溶液,19.4mL氮溶液,0.5mL维他命溶液,pH4.5;矿质溶液为两种,一种为含Mn2+矿质溶液,一种为不含Mn2+矿质溶液;当采用B、C或D培养条件进行诱导培养时,前期菌种培养采用的LNAS液体培养基中的矿质溶液为含Mn2+矿质溶液,其组成配方为(1000mL):0.087g柠檬酸铁,0.1gZnSO4·7H2O,0.45gMnSO4·H2O,0.1gCoCl2·6H2O,0.01gCuSO4·5H2O;当采取A培养条件进行诱导培养时,前期菌种培养采用的LNAS液体培养基中的矿质溶液为不含Mn2+矿质溶液,其组成配方为(1000mL):0.087g柠檬酸铁,0.1gZnSO4·7H2O,0.1gCoCl2·6H2O,0.01gCuSO4·5H2O;氮溶液的组成配方为(1000mL):4.0gL-天门冬素,2.0gNH4NO3;维他命溶液的组成配方为(1000mL):0.002gvitaminH,0.002gvitaminM,0.005gvitaminB1,0.005gvitaminB2,0.01gvitaminB6,0.0001gvitaminB12,0.005g烟酸,0.005g泛酸钙,0.005g对氨基苯甲酸,0.005gDL-6,8-硫辛酸。实施例3本实施例说明白腐菌树舌基因组总DNA的提取:(1)取新鲜白腐菌树舌菌丝20mg,放入研钵中加入液氮充分碾磨,加入400μl缓冲液FP1和6μl的RNaseA(10mg/mL),旋涡振荡1min,室温放置10min;(2)加入130μl缓冲液FP2,充分混匀,涡旋振荡15min,12000rpm离心5min,将上清转移至新的离心管中;(3)向上清液中加入0.7倍体积的异丙醇,充分混匀,此时出现絮状基因组DNA,12000rpm离心2min,弃上清,保留沉淀;(4)加入700μl70%乙醇,涡旋振荡5s,12000rpm离心2min,弃上清;(5)重复步骤4;(6)开盖倒置,室温8min,彻底晾干残余的乙醇,加入适量洗脱缓冲液TE,65℃水浴50min溶解DNA,期间颠倒混匀助溶,最终得到DNA溶液;(7)采用1%琼脂糖凝胶电泳检测完整性,图3为其相应电泳检测图,检测后的基因组DNA置于-20℃冰箱保存。实施例4本实施例说明利用白腐菌树舌基因组总DNA获取Ga-MnP目的基因片段:(1)提取白腐菌树舌基因组总DNA后,对比已报道的其它白腐菌锰过氧化物酶同源基因序列,利用软件ClustalX2.0进行相似序列比对分析,界定该类基因的保守序列LTFHDAI和HTVGSQN;(2)设计简并引物Ga-MnPF(5'-CTGACCTTCCATGACGCTAT-3)、Ga-MnPR(5'-GTATGGCAGCCAAGCGTAGGG-3'),通过简并PCR反应获得Ga-MnP目的基因片段;其中,简并PCR反应体系如表1所示,反应条件为:94℃预变性3min,94℃30S,55℃30S,72℃2min,30个循环,72℃10min,PCR产物经1%的琼脂糖凝胶电泳检测,如图4所示。表1简并PCR反应体系10×PCRBuffer5μldNTPMixture(10mMeach)2μlGa-MnPF(10μM)1μlGa-MnPR(10μM)1μlTaKaRaExTaqTMHS(5U/μl)0.5μl上述反应液≤5μlRNaseFreedH2O补足至50μl实施例5本实施例说明白腐菌树舌基因组总RNA的提取:(1)取小于100mg的白腐菌树舌菌丝放入研钵中,加液氮研磨成糊状,用小药匙刮研钵壁上研磨好的组织并转移到离心管内;(2)将研磨好的白腐菌树舌菌丝放入离心管后,立即添加600μlBufferRB/β-巯基乙醇;(3)用枪头将裂解液迅速移入到放置在2mL离心管的Homogenizationspincolumn中,室温13000×g离心5min;(4)转移上清液到一个新的离心管内,加入0.5倍体积的无水乙醇;(5)取一个HiBindRNA小量柱,并把柱子装在一个收集管内,转移混合液至柱子中,轻轻的盖上管帽,室温下10000×g离心30s,弃去收集管中的滤液,并把柱子重新装在收集管中;(6)把柱子重新放入新的收集管中,添加300μlRNA洗涤Buffer1到柱中,室温10000×g离心1min;(7)将75μlDNase1消化处理混合剂立即准确加入到每个HiBidRNA柱膜的中央,室温下(25-30℃)放置15min;(8)将柱子放置在新的收集管上,添加500μlRNA洗涤Buffer1,室温静置5min,室温下,10000×g离心1min,弃滤液,重新使用一个收集管;(9)将柱子放置在同样的收集管上,添加500μlRNA洗涤Buffer2,室温下,10000×g离心1min,弃滤液,再次使用收集管;(10)用500μlRNA洗涤Buffer2第二次洗涤柱子,重复上一步骤,室温下,10000×g离心1min,弃滤液,然后使用新的收集管,室温下,10000×g离心1min,以甩干柱子的基质;(11)洗脱RNA,将柱子转移到新的离心管上并且用80μl的DEPC水洗涤RNA,电泳检测,再将提取的白腐菌树舌总RNA溶液贮存于-80℃保存备用,其中电泳结果如图5所示。实施例6本实施例说明利用白腐菌树舌基因组总RNA结合反转录法合成cDNA第一链:取1-2μg总RNA,加入ddH2O至9.5ul,75℃变性5min,冰浴5min,稍微离心;配制20ul的逆转录RT-PCR反应体系、进行反转录反应:(1)在Microtube管中配制如表2所示的混合液;表2混合液(2)在PCR仪上进行变性、退火反应:65℃5min;(3)离心数秒钟使模板RNA/引物等的混合液聚集于Microtube管底部;(4)在上述Microtube管中配制如表3所示的反转录反应液;表3反转录反应液上述变性、退火后反应液10μl5×PrimeScriptTMBuffer4μlRNaseInhibitor(40U/μl)0.5μlPrimeScriptTMRTase(for2Step)0.5μlRNaseFreedH2O5μl总体积20μl(5)在PCR仪上按下列条件进行反转录反应:42℃30min,95℃5min;接着,进行进一步地PCR反应获取Ga-MnP目的基因,PCR反应液配比如表4,反应条件为:94℃预变性3min,94℃30S,55℃30S,72℃2min,30个循环,72℃10min;PCR产物经1%的琼脂糖凝胶电泳检测,检测结果如图6所示。表4PCR反应液配比注:Ga-MnPF引物:5'-CTGACCTTCCATGACGCTAT-3'Ga-MnPR引物:5'-GTATGGCAGCCAAGCGTAGGG-3'实施例7本实施例说明目的基因PCR产物凝胶回收并与T载体连接:(1)将带有目的片段的凝胶块转移至离心管,加入等体积的BindingBuffer(XP2),于55-60℃水浴中温浴7min或至凝胶完全融化,每2-3min振荡或涡旋混合物;(2)取一个干净的HiBindDNAMini柱子装在一个干净的收集管内,将DNA/熔胶液全部转移至柱子中,室温下10000×g离心1min,弃收集管中的滤液,将柱子套回收集管;(3)将柱子套回收集管,转移300μlBindingBuffer(XP2)至柱子中,室温下,10,000×g离心1min;(4)弃收集管中的滤液,将柱子套回收集管内收集管,转移700μlSPWWashbuffer(已用无水乙醇稀释)至柱子中,10000×g离心1min;(5)重复步骤4;(6)弃收集管中的滤液,将柱子套回收集管内收集管,室温下,13000xg离心2min以甩干柱子基质残余的液体;(7)把柱子装在一个干凈的离心管上,加入30-50μl的ElutionBuffer到柱基质上,室温放置1min,13000×g离心1min,洗脱DNA;回收产物经1%的琼脂糖凝胶电泳检测;(8)在微量离心管中配制如表5的连T混合液:表5连T混合液在上述混合液中加入(等量)SolutionⅠ5μl,全量10μl,16℃连接过夜。实施例8本实施例说明连接后的产物进行转化:(1)吸取全量10μl连接产物,加入到100μl感受态细胞悬浮液中,冰浴30min;(2)42℃水浴加热45s,冰上放置1min;(3)加入890μl的SOC培养基,37℃振荡培养60min;(4)在含有X-Gal、IPTG、Amp的LB琼脂平板培养基上培养,形成单菌落;(5)挑选白色菌落至含有Amp抗性的LB液体培养基中,37℃、180-200r/min振荡培养10h左右,提取白色单菌落进行PCR鉴定。实施例9本实施例说明利用利用RACE技术获得基因全长序列及该基因Ga-MnP的扩增、连载体及测序:(1)3′RACE的反转录反应:首先用ReverseTranscriptaseM-MLV(RNaseH-)将总RNA反转录合成单链cDNA,再使用TaKaRaLATaq酶,以反转录的cDNA为模板,进行嵌套PCR,扩增目的基因的3′端。①以TotalRNA为模板,使用3′RACEAdaptor引物进行反转录反应,合成单链cDNA。反应中总RNA的用量范围为:50ng-1μg,反应体系如表6;表63′反转录反应合成单链cDNA的反应体系总RNAXμl3′RACEAdaptor(5μM)1μl5×M-MLVBuffer2μldNTPMixture(10mMeach)1μlRNaseInhibitor(40U/μl)0.25μlReverseTranscriptaseM-MLV(RNaseH-)(200U/μl)0.25μlRNaseFreedH2O5.5~Xμl总体积10μl注:3’RACE接头引物(AdaptorPrimer):5'-GCTGTCAACGATACGCTACGTAACGGCATGACAGTG(T)-3'②反应条件:42℃,60min70℃,15min。反应结束后可以进行下一步实验或将反应液保存于-20℃。(2)3′RACE的套式PCR反应:①使用TaKaRaLATaq进行嵌套PCR反应,使用上游外侧特异性引物(GSP)和3′RACEOuterPrimer进行1stPCR反应,GeneSpecificOuterPrimer使用3′RACE基因外侧特异性引物RAP-1,特异性引物是根据已扩增出的目的基因片段设计,反应体系如表7:表73′RACE的套式PCR反应反应体系1注:3’RACE外侧引物(OuterPrimer):5'-CTGACCTTCCATGACGCTATC-3'3’RACE特异性引物GSP:5'-TTCCACGACGCAATTGGTTTC-3'②反应条件:94℃预变性3min,94℃30S,55℃30S,72℃2min,20个循环,72℃10min。PCR产物经1%的琼脂糖凝胶电泳检测后若未获得目的基因片段可进行内侧PCR反应。③如果1stPCR反应未能得到目的产物,再使用上游内侧特异性引物(GSP2)和3′RACEInnerPrimer进行2ndPCR反应。InnerPCR反应中GeneSpecificInnerPrimer为3′RACE基因内侧特异性引物(GeneSpecificInnerPrimer),进一步扩增出目的基因,反应体系如表8;表83′RACE的套式PCR反应反应体系21stPCR产物1μldNTPMixture(2.5mMeach)8μl10×LAPCRBufferII(Mg2+Free)5μlMgCl2(25mM)5μlTaKaRaLATaq(5U/μl)0.5μl3’RACEInnerPrimer(10μM)2μlGeneSpecificInnerPrimer(10μM)2μldH2O26.5μl总体积50μl注:3’RACE内侧引物(InnerPrimer):5'-CCATACCGTCGGTTCGCAGAAC-3'3’RACE内侧特异性引物(GeneSpecificInnerPrimer):5'-CAGCATTCTTTCTCGCTTCACT-3'④反应条件:94℃预变性3min,94℃30S,55℃30S,72℃2min,30个循环,72℃10min,PCR产物经1%的琼脂糖凝胶电泳检测,结果如图7所示;(3)3′RACE产物的回收、T载体克隆及测序PCR产物凝胶回收、T载体克隆、质粒提取及PCR验证,步骤如同其上,本步骤T克隆所用感受态细胞为E.coliJM109,经过验证后菌液,送测序公司测序;(4)5′RACE的cDNA第一链合成以mRNA为模板,反转录合成cDNA的第一条链,然后用PCR技术扩增出从某个特定位点到5’端之间的未知核苷酸序列,以获得的MnP基因序列为基础,设计5’RACE特异性引物,用5’RACE技术克隆出MnP目的基因的5′端cDNA序列,可以拼接出MnP全长cDNA序列;①在PCR管中加入如表9的试剂:表95′RACE的cDNA第一链合成的试剂1总RNA1~3μl5′-CDSprimerA1μlSMARTⅡAOligo1μlRNaseFreedH2O补足至5μl②混合并在小型离心机上短暂离心,70℃下温育2min,迅速在冰上冷却2min,短暂离心使成份聚集管底;③在上述反应管中再加入如表10的试剂:表105′RACE的cDNA第一链合成的试剂2上述反应液5μl5×First-StrandBuffer2μlDTT(20mM)1μldNTPMix(10mM)1μlMMLVReverseTranscriptase1μl总体积10μl④小心混合管内组分,短暂离心使成份聚集管底,恒温箱中42℃下温育1.5h;⑤使用Tricine-EDTA缓冲液来稀释反应产物,72℃下加热管7min,所得产物可直接进行下一步反应,也可以在–20℃下保存,保存期不超过3个月;(5)5′RACE的cDNA末端的快速扩增(RACE)PCR反应使用AdvantagePCR试剂,其中GSP为5′RACE特异性引物是根据已扩增出的目的基因所设计,反应体系(50μl)如表11:表115′RACE的cDNA末端的快速扩增的试剂PCRGradeWater34.5μl10×Advantage2PCRBuffer5μldNTPMix(10mM)1μl5′RACE-ReadycDNA2.5μlUPM(10×)5μlGSP1(10μM)1μl50×Advantage2polymeraseMix1μl总体积50μl注:5’RACE特异性引物:5'-GCGCTTGACGTTACCCCCGGGAG-3'反应条件为:94℃预变性3min,94℃30S,68℃30S,72℃3min,30个循环,72℃10min;PCR产物经1%的琼脂糖凝胶电泳检测,结果如图8所示;(6)5′RACE产物的回收、T载体克隆及测序PCR产物凝胶回收、20-T载体的连接、转化大肠杆菌感受态细胞、质粒的提取、PCR验证阳性转化子及测序,步骤同上;(7)白腐菌树舌MnP全长cDNA基因的扩增使用RACE方法扩增出MnP的cDNA是无内含子的全长基因,分别以起始密码子ATG和终止密码子TAA附近碱基设计上下游引物,反应体系(50μl)如表12:表12全长cDNA基因的扩增反应体系10×LAPCRBuffer5μldNTPMixture(10mMeach)2μl上游引物1(10μM)1μl下游引物2(10μM)1μlTaKaRaLATaqTMHS(5U/μl)0.5μl上述反转录反应液≤5μlRNaseFreedH2O补足至50μl注:上游引物1:5'-ATGTTCTCAAAAGTCTTCCTCTC-3'下游引物2:5'-GGGCGACACGGGAACTTGAGGACT-3'反应条件为:94℃预变性3min,94℃30S,58℃30S,72℃3min,30个循环,72℃10min;PCR产物经1%的琼脂糖凝胶电泳检测,结果如图9所示;(9)MnP全基因产物的回收、T载体克隆及测序PCR产物凝胶回收、20-T载体的连接、转化大肠杆菌感受态细胞、质粒的提取、PCR验证阳性转化子及测序,步骤同上。以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1