噁二唑羧酸金属配合物及其制备方法和应用与流程
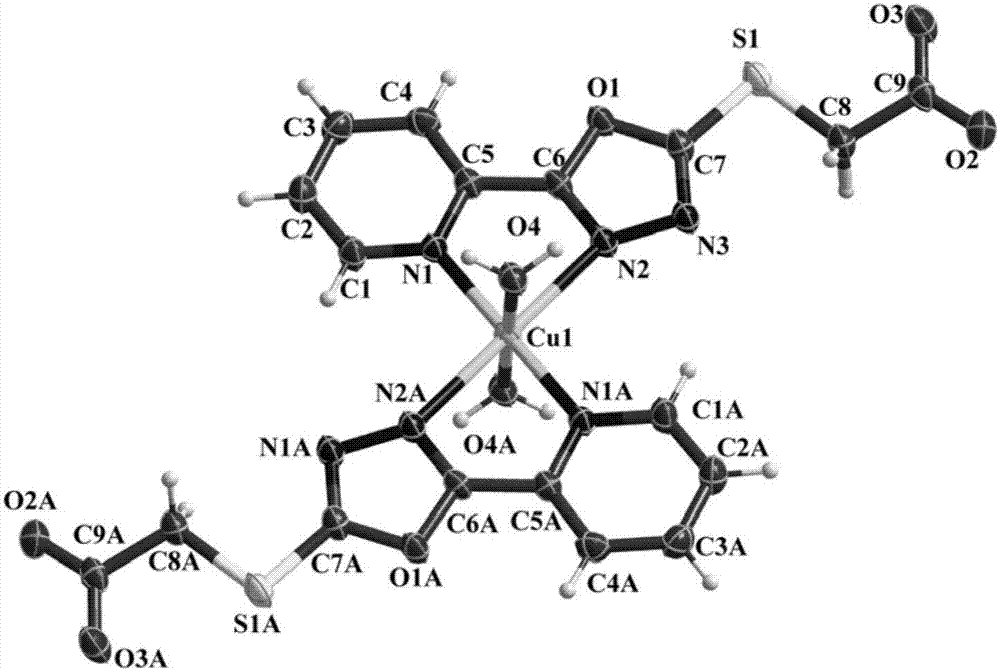
本发明属于生物功能羧酸配合物
技术领域:
,具体涉及一种噁二唑羧酸金属配合物及其制备方法和应用。
背景技术:
:美国科学家rosenberg于1969年首次报道了顺式-二氯二氨合铂(ⅱ)具有抗肿瘤活性后,开启了金属配合物在医药领域应用的新篇章。羧基(-cooh)中两个氧原子含有四对未共用电子对,它们都可用来与金属离子结合。与金属配位时能以单齿、螯合或桥连形式配位,全部或部分去质子化从而展现出不同的配位几何,是配位化学中最常用的配位基团。此外,羧基形成非共价键的能力也很强,根据去质子羧基的数目能分别作为氢键的授体和受体,因此易与靶点蛋白活性位点上的氨基酸形成较强的氢键相互作用。羧基配合物一般都具有较好的稳定性,可以降低金属离子在生物体内被释放的可能性,此外羧基本身的电负性,可以降低配合物整体的电性,使之较容易的透过细胞膜。lei,n.;ren.q.l.;liu,y.p.;li,j.;cong,p.;qin,j.;zhu,h.l.j.mol.struct.2014,1067,220-224;hussein,b.h.m.;azab,h.a.;el-azab,m.f.;el-falouji,a.i.eur.j.med.chem.2012,51,99-109;yang,z.m.;zhu,h.;sun,j.;qian,s.s.;can,m.n.;zhu,h.l.j.coord.chem.2013,66,2736-2746,上述文献公开了羧酸配合物在药物领域的应用。含1,3,4-噁二唑骨架的分子具有非常广泛的生物活性,如抗真菌、抗细菌、抗植物病毒、抗炎、抗焦虑及抗结核等,该类化合物的分子设计和合成工艺研究一直是人们关注的热点。sun,j.;cao,n.;zhang,x.m.;yang,y.s.;zhang,y.b.;wang,x.m.;zhu,h.l.bioorg.med.chem.2011,19,4895-4902;manjunatha,k.;poojary,b.;prajwal,l.l.;fernandes,j.;kumari,n.s.eur.j.med.chem.2010,45,5225-5233.;sun,j.;zhu,h.;yang,z.m.;zhu,h.l.eur.j.med.chem.2013,60,23-28,上述文献公开了噁二唑衍生物在药物领域的应用。利用活性骨架拼接原理,合成噁二唑羧酸衍生物具有较好的应用前景,对此进行深入研究具有一定的理论和实际价值,尤其是合成噁二唑羧酸金属配合物并对它们的生物活性进行系统的研究具有十分重要的意义。技术实现要素:本发明的目的是提供一种噁二唑羧酸金属配合物,具有合成简单,便于提纯,产率较高,在空气中稳定性好的特点;本发明同时提供其制备方法和应用。本发明所述的噁二唑羧酸金属配合物,是单核化合物,其结构式如下:其中:m为ag或cu;m为ag时,r1=cooh,无r2,r3=h2o;m为cu时,r1=coo-,r2=h2o,无r3。所述的噁二唑羧酸金属配合物的制备方法,包括以下步骤:(1)将2-吡啶甲酰肼与氢氧化钾、甲醇混合,冰水浴下滴加二硫化碳,滴加完毕回流反应,反应结束,旋干溶剂,加水过滤,滤液酸化,得到白色固体5-(2-吡啶)-1,3,4-噁二唑-2-硫醇;(2)将5-(2-吡啶)-1,3,4-噁二唑-2-硫醇与氢氧化钾、甲醇混合搅拌至澄清,加入溴乙酸常温反应,反应结束过滤,得到白色固体5-(2-吡啶)-1,3,4-噁二唑-2-巯基乙酸;(3)将5-(2-吡啶)-1,3,4-噁二唑-2-巯基乙酸溶于丙酮中,硝酸银或硝酸铜溶于水中,将上述两种溶液混合后避光静置,得到噁二唑羧酸金属配合物。其中:步骤(1)中2-吡啶甲酰肼、氢氧化钾、二硫化碳、甲醇的用量比例为1:1.5:10:7,其中,2-吡啶甲酰肼、氢氧化钾、二硫化碳以毫摩尔数计,甲醇以毫升计。步骤(1)中滴加二硫化碳的时间为15-20分钟。步骤(1)中回流时间为5-6小时。步骤(2)中5-(2-吡啶)-1,3,4-噁二唑-2-硫醇、氢氧化钾、溴乙酸、甲醇的用量比例为1:1:1:5,其中,5-(2-吡啶)-1,3,4-噁二唑-2-硫醇、氢氧化钾、溴乙酸以毫摩尔数计,甲醇以毫升计。步骤(2)中常温反应时间为6-7小时。步骤(3)中5-(2-吡啶)-1,3,4-噁二唑-2-巯基乙酸、硝酸银或硝酸铜、丙酮、水的用量比例为1:1:125:125,其中,5-(2-吡啶)-1,3,4-噁二唑-2-巯基乙酸、硝酸银或硝酸铜以毫摩尔数计,丙酮和水以毫升计。步骤(3)中静置时间为3-5天。本发明所述的噁二唑羧酸金属配合物可用于脲酶抑制剂的制备。本发明的反应方程式如下:m为ag时,x=1,r1=cooh,无r2,r3=h2o;m为cu时,x=2,r1=coo-,r2=h2o,无r3。本发明用红外、元素分析(c,h,n)、x-射线单晶衍射表征并证实了噁二唑羧酸配合物的结构。本发明的噁二唑羧酸银、铜配合物具有很强的脲酶抑制活性,半数抑制浓度分别为0.74μmol/l,1.28μmol/l。本发明的有益效果如下:本发明所述的噁二唑羧酸金属配合物合成简单,便于提纯,产率较高,在空气中稳定性好;其制备方法简单易行;本发明的噁二唑羧酸银、铜配合物可用于脲酶抑制剂的制备,具有很强的脲酶抑制活性,抑制能力优于阳性对照药物。附图说明图1为本发明实施例1的单晶结构图;图2为本发明实施例2的单晶结构图。具体实施方式以下结合实施例对本发明做进一步描述。实施例15-(2-吡啶)-1,3,4-噁二唑-2-巯基乙酸合银的制备方法如下:(1)将2-吡啶甲酰肼(1.38g,10mmol),氢氧化钾(0.84g,15mmol)溶于70ml甲醇中,冰水浴中滴加二硫化碳(6.04ml,100mmol),20分钟内滴完。滴加完二硫化碳后回流反应6h,旋干溶剂,加水溶解,滤液加盐酸酸化,过滤,得白色固体5-(2-吡啶)-1,3,4-噁二唑-2-硫醇1.48g,产率:83%;化合物经红外、元素分析进行了验证,结果表明结构正确,数据如下:红外(kbr,cm-1):3391,1862,1612,1558,1569,1458,1381,1342,1275,1175,990,786,698(m)。元素分析结果:计算值(%):c,46.92;h,2.81;n,23.45;实测值(%):c,47.06;h,2.79;n,23.54%。(2)将5-(2-吡啶)-1,3,4-噁二唑-2-硫醇(0.90g,5mmol),氢氧化钾(0.28g,5mmol)溶于25ml甲醇中,常温搅拌至澄清后,加入溴乙酸(0.69g,5mmol)常温搅拌6小时,产生的沉淀过滤,得白色固体5-(2-吡啶)-1,3,4-噁二唑-2-巯基乙酸0.88g,产率:74%;化合物经红外、元素分析进行了验证,结果表明结构正确,数据如下:红外(kbr,cm-1):3090,2983,2934,2784,2663,2539,1720,1595,1479,1463,1377,1307,1289,1201,1170,1088,960,799,751,703,633,513。元素分析结果:计算值(%):c,45.56;h,2.97;n,17.71;实测值(%):c,45.74;h,2.95;n,17.76。(3)将5-(2-吡啶)-1,3,4-噁二唑-2-巯基乙酸(9.4mg,0.04mmol)溶于5ml丙酮溶液中,然后将其缓慢滴入含有硝酸银(6.8mg,0.04mmol)5ml水溶液中,避光静置5天,出现7.5mg淡黄色晶体,过滤,并用甲醇洗涤三次,置于无水氯化钙干燥器内干燥得产品,产率61%。产品经红外、元素分析进行了验证,结果表明结构正确,数据如下:红外(kbr,cm-1):3382,3090,2933,1720,1599,1462,1384,1307,1200,1170,1087,1008,960,915,797,747,702,631。元素分析结果:计算值(%):c,35.47;h,2.81;n,13.79;实测值(%):c,35.57;h,2.79;n,13.84。5-(2-吡啶)-1,3,4-噁二唑-2-巯基乙酸合银的结构式如下:上述配合物分子式、性状、产率等性质见表1。实施例25-(2-吡啶)-1,3,4-噁二唑-2-巯基乙酸合铜的制备方法如下:将5-(2-吡啶)-1,3,4-噁二唑-2-巯基乙酸(9.4mg,0.04mmol)溶于5ml丙酮溶液中,然后将其缓慢滴入含有硝酸铜(9.6mg,0.04mmol)5ml水溶液中,避光静置3天,出现7.7mg墨绿色晶体,过滤,并用甲醇洗涤三次,置于无水氯化钙干燥器内干燥得产品,产率67%。产品经红外、元素分析进行了验证,结果表明结构正确,数据如下:红外(kbr,cm-1):3425,3081,1614,1556,1448,1379,1228,1192,1122,1089,1031,913,766,699,672,426。元素分析结果:计算值(%):c,37.79;h,2.82;n,14.69;实测值(%):c,37.86;h,2.80;n,14.74。5-(2-吡啶)-1,3,4-噁二唑-2-巯基乙酸合铜的结构式如下:上述配合物分子式、性状、产率等性质见表1。表1两种噁二唑羧酸金属配合物序号配合物分子式性状产率(%)实施例1c18h17n6o8s2ag淡黄色晶体61实施例2c18h16n6o8s2cu墨绿色晶体67以下对实施例1-2得到的配合物进行检测。1、噁二唑羧酸金属配合物的单晶结构表征。测试仪器为brukersmartapexccd单晶衍射仪,以mo-kα(λ=0.071073nm)射线,在25℃时用ω/2θ的扫描模式进行测试。数据通过saint修正,lorentz修正和极化效应的消除得到。吸收修正使用bruker的sadabs补充。用shelxl-97直接解出了分子结构。金属原子及其周围相连原子的位置用直接e-maps的方法测出,其他非氢原子通过傅立叶变换,最小二乘法修正逐步确定其精细结构。氢原子则最后确定于计算所得的位置,并有统一的uiso值(附图1-2)。检测结果如表2所示。表2实施例1和实施例2的单晶衍射数据表ar1=σ||c|-|fc||/σfo|.bwr2=[σw(fo2-fc2)2/σw(fo2)]1/2。2、本发明噁二唑羧酸金属配合物的脲酶抑制作用检测。测试方法及步骤:25μl巨豆脲酶(10ku/l)加25μl样品溶液(不同浓度,dmso/h2o=1:1溶解)于96孔培养板中37℃培养1小时,再加入0.2ml100mmhepes缓冲液(ph=6.8)(内含:500mm尿素和0.002%酚红),用乙酰氧肟酸作为阳性对照,计算缓冲液ph由6.8升至7.7时所用时间,并在570nm波长下用酶标仪检测od值,计算半数抑制浓度ic50。半数抑制浓度越小,表明化合物的脲酶抑制活性越好,实施例1-2和阳性对照的脲酶抑制活性测试结果如表3。表3脲酶抑制活性测试结果表(μmol/l)实施例中检测所用仪器为:perkin-elmer240c型元素分析仪,vector22bruker分光光度计(400-4000cm-1),brukersmartapexccd单晶衍射仪以及biorad550型酶标仪等。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种羰基锝-99m标记的美法仑配合物及其制备方法与用图
- 一种2-羰基-3-苯基丙酸苯甲酰腙二苯基锡配合物及其制备方法和应用
- 一种新型金属有机配合物纤维及其衍生多孔碳纤维的制备方法
- 包含新的配体结构的金属配合物的制作方法
- 一种2-羰基-3-苯基丙酸苯甲酰腙二(2,4-二氯苄基)锡配合物及其制备方法和应用
- 一种2-羰基-2-苯基乙酸苯甲酰腙二丁基锡配合物及其制备方法和应用
- 一种2-羰基-3-苯基丙酸水杨酰腙二苯基锡配合物及其制备方法和应用
- 一种2-羰基-3-苯基丙酸苯甲酰腙二(2,4-二氯苄基)锡配合物及其制备方法和应用
- 一种2-羰基-2-苯基乙酸苯甲酰腙二丁基锡配合物及其制备方法和应用
- 一种2-羰基-3-苯基丙酸水杨酰腙二苯基锡配合物及其制备方法和应用