一种四氢萘啶并四氢喹唑啉衍生物及其合成方法和应用与流程
本发明属于有机合成技术领域,具体涉及一种四氢萘啶并四氢喹唑啉衍生物及其合成方法和应用。
背景技术:
杂环化合物是一类非常重要的有机化合物,在过去的近十年当中,杂环化学
近些年,合成含有四氢萘啶类或四氢喹唑啉类化合物的方法陆续报道,ippolitoantonini(ippolitoantonini,paolopolucci,lloydr.kelland,ernestomenta,nicolettapescalli,andsantemartelli.j.med.chem.1999,42,2535-2541)团队通过四步法合成只含有四氢喹唑啉结构的衍生物。这些传统的方法需要较为复杂的条件——合成步骤繁琐,需要添加强酸性条件、较高的温度、很长的反应时间等。然而合成四氢萘啶并四氢喹唑啉结构的稠环骨架化合物几乎没有报道。
技术实现要素:
为了解决以上现有技术的缺点和不足之处,本发明的首要目的在于提供一种四氢萘啶并四氢喹唑啉衍生物。
本发明的另一目的在于提供一种快速、高效的四氢萘啶并四氢喹唑啉衍生物的合成方法。
本发明的再一目的在于提供上述四氢萘啶并四氢喹唑啉衍生物的用途。
本发明目的通过以下技术方案实现:
一种四氢萘啶并四氢喹唑啉衍生物,其分子式如下式i:
其中,所述的r1为脂肪类基团,r2为脂肪类基团或者芳香类基团。
优选的,r1为甲基、甲氧基、硝基、腈基、三氟甲基、酯基、卤素取代基或氢等;r2为烷基、苯基、萘基、呋喃基或噻吩基等。
一种四氢萘啶并四氢喹唑啉衍生物的合成方法,包括以下步骤:
(1)将2-氨基-3-吡啶甲醛和苯甲酰乙腈加入反应容器中,2-氨基-3-吡啶甲醛和苯甲酰乙腈摩尔比为5:6,然后添加叔丁醇钾和乙醇,60℃反应5小时,分离提纯得到2-苯基3-腈基1,8萘啶;
(2)在反应器中,加入四氢喹啉和2-苯基3-腈基1,8萘啶,四氢喹啉和2-苯基3-腈基1,8萘啶的摩尔比为1∶1~2,加入四氢喹啉质量1~5%的金属催化剂,加入溶剂,在80℃~160℃下搅拌反应1~24小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到1,2,3,4-四氢萘啶类化合物;
(3)在反应器中,加入1,2,3,4-四氢萘啶类化合物与醛,1,2,3,4-四氢萘啶类化合物与醛的摩尔比为1∶1~3,加入溶剂,在0℃~100℃下搅拌反应1~12小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到四氢萘啶并四氢喹唑啉衍生物。
步骤(2)所述的金属催化剂为醋酸铜、硫酸铜、氯化铁、醋酸钯、双三苯基膦二氯化钯、三氯化铱、1,5-环辛二烯氯化铱二聚体、二氯(五甲基环戊二烯基)合铱(iii)二聚体、十二羰基三钌和环戊二烯基双(三苯基膦)氯化钌(ii)中的一种或两种以上的混合。
本发明所述的室温是指20-30℃。
步骤(2)所述的溶剂为乙醇、叔戊醇、异丙醇、1,4-二氧六环、n,n-二甲基甲酰胺、二甲基亚砜、甲苯、对二甲苯和水中的一种或两种以上的混合;其加入量与四氢喹啉的比例优选为四氢喹啉:溶剂=0.5mmol:1~3ml。
步骤(3)所述的醛为脂肪醛或者芳醛。
步骤(3)所述的醛为甲醛、乙醛、丙醛、苯甲醛、对甲基苯甲醛、对甲氧基苯甲醛、对溴苯甲醛、对氯苯甲醛、对硝基苯甲醛和对酯基苯甲醛。
步骤(3)所述的溶剂为甲酸、磷酸、冰醋酸、甲苯、甲醇和水中的一种或两种以上的混合;其加入量与1,2,3,4-1,2,3,4-四氢萘啶类化合物之间的比例优选为1,2,3,4-1,2,3,4-四氢萘啶类化合物:溶剂=0.3mmol:1-2ml。
步骤(2)、(3)所述的反应器优选schlenk管(史兰克管),氮气条件下。
步骤(2)、(3)所述的柱层析提纯所用的洗脱液为石油醚:二氯甲烷:乙酸乙酯的体积比为(0.5~50):(0~20):1的混合溶剂。
上述合成方法所涉及的部分反应方程式如下:
其中,r1为脂肪类基团,优选为甲基、甲氧基、硝基、腈基、三氟甲基、酯基、卤素取代基或氢等;r2为脂肪类基团或者芳香类基团,优选为烷基、苯基、萘基、呋喃基或噻吩基等。
上述四氢萘啶并四氢喹唑啉衍生物表现出潜在的药物活性,比如前列腺素e1受体拮抗剂、抗菌药物、酪氨酸激酶抑制作用、抗肿瘤、抗癫痫药和抗惊厥药,因此可用于制备前列腺素e1受体拮抗剂、抗菌药物、酪氨酸激酶抑制剂、抗肿瘤药物、抗癫痫药或抗惊厥药。
与现有技术相比,本发明具有以下优点及有益效果:
本发明以四氢喹啉与萘啶为原料一步合成四氢萘啶并四氢喹唑啉类化合物,具有合成步骤简单、操作安全、原料无毒且价廉易得、合成方法对功能团兼容性好、原子经济性高的优点。得到的四氢萘啶并四氢喹唑啉类产物结构新颖,可进一步开发其生物活性。
附图说明
图1和图2分别为实施例1所得产物1a的氢谱图和碳谱图;
图3和图4分别为实施例1所得产物1b的氢谱图和碳谱图;
图5和图6分别为实施例2所得产物2a的氢谱图和碳谱图;
图7和图8分别为实施例2所得产物2b的氢谱图和碳谱图;
图9和图10分别为实施例3所得产物3b的氢谱图和碳谱图;
图11和图12分别为实施例4所得产物4b的氢谱图和碳谱图;
图13和图14分别为实施例5所得产物5b的氢谱图和碳谱图。
具体实施方式
下面结合实施例和附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
以下实施例中使用的2-苯基3-腈基1,8萘啶通过以下步骤制得:
圆底烧瓶里加入2-氨基-3-吡啶甲醛(反应方程式中的a,5mmol)和苯甲酰乙腈(反应方程式中的b,6mmol),然后添加叔丁醇钾(1mmol)、5ml乙醇做溶剂,60℃反应5小时,分离提纯得到2-苯基3-腈基1,8萘啶。
实施例1
(1)在反应器中,加入0.5毫摩尔四氢喹啉和0.3毫摩尔2-苯基3-腈基1,8萘啶,0.01毫摩尔1,5-环辛二烯氯化铱二聚体,1.5ml甲苯,在100℃下搅拌反应16小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到四氢萘啶并四氢喹唑啉化合物1a;
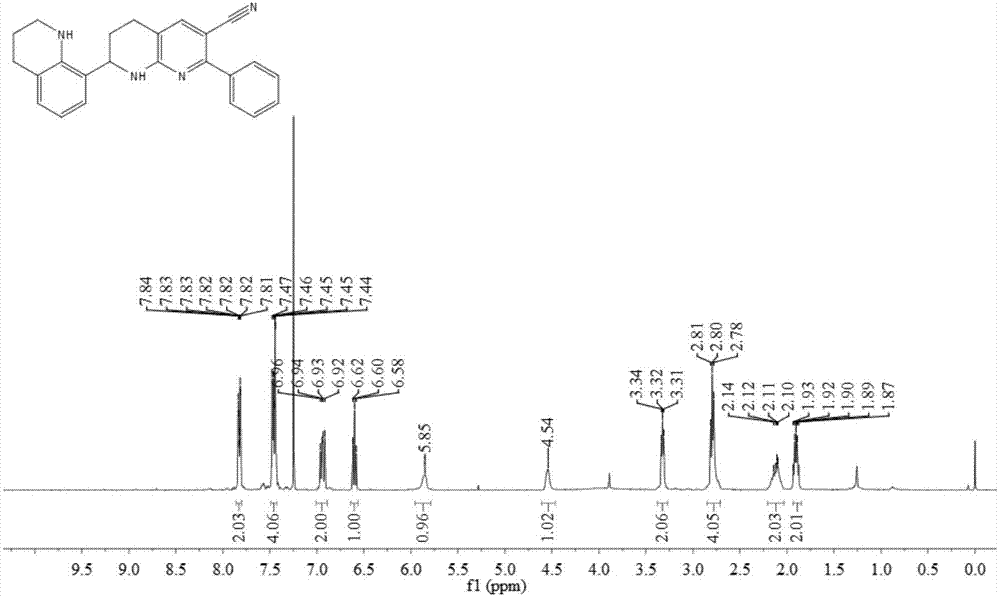
所得产物1a的氢谱图及碳谱图分别如图1和图2所示,结构表征数据如下:
1hnmr(400mhz,cdcl3):δ7.86–7.80(m,2h),7.49–7.43(m,4h),6.94(dd,j=11.3,7.6hz,2h),6.60(t,j=7.5hz,1h),5.85(s,1h),4.61–4.47(m,1h),3.38–3.27(m,2h),2.85–2.71(m,4h),2.20–2.03(m,2h),1.93–1.85(m,2h);
13cnmr(101mhz,cdcl3):δ159.65,157.72,141.51,139.85,137.93,129.53,129.18,128.67,128.43,124.62,122.25,119.85,116.56,114.71,94.25,51.86,42.16,27.64,24.90,24.47,21.76;
ir(kbr):3407,3057,2940,2844,2211,1712,1602,1507,1435,1279,1188,1123,918cm-1;
yellowsolid,m.p:189-191℃;
hrms(esi):calcd.forc24h22n4[m+h]+:367.1917;found:367.1922。
根据以上数据推断所得产物1a的结构如下式所示:
(2)在反应器中,加入0.1毫摩尔1a与0.2毫摩尔苯甲醛,加入1ml冰醋酸,在60℃下搅拌反应5小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到四氢萘啶并四氢喹唑啉类化合物1b。
所得产物1b的氢谱图及碳谱图分别如图3和图4所示,结构表征数据如下:
1hnmr(400mhz,cdcl3):δ7.98(d,j=6.8hz,2h),7.57–7.47(m,4h),7.45(s,1h),7.37(d,j=6.8hz,2h),7.35–7.27(m,3h),6.94(d,j=7.3hz,1h),6.89(d,j=7.6hz,1h),6.61(t,j=7.5hz,1h),4.54(t,j=5.0hz,1h),3.51–3.35(m,2h),2.93–2.72(m,4h),2.35–2.22(m,2h),2.13–1.96(m,2h);
13cnmr(101mhz,cdcl3):δ158.67,155.60,140.39,140.21,139.90,138.24,129.55,128.73,128.70,128.42,128.08,127.91,126.99,122.27,121.55,120.76,119.74,116.62,115.86,94.84,69.05,50.03,48.34,27.69,24.70,23.92,21.55;
ir(kbr):3059,2924,2336,2213,1730,1710,1598,1493,1314,1270,1016,699cm-1;
brownishsolid,m.p:101-103℃;
hrms(esi):calcd.forc31h21n4[m+h]+:455.2230;found:455.2233。
根据以上数据推断所得产物1b的结构如下式所示:
实施例2
(1)在反应器中,加入0.6毫摩尔6-甲基四氢喹啉和0.3毫摩尔2-苯基3-腈基1,8萘啶,0.05毫摩尔二氯(五甲基环戊二烯基)合铱(iii)二聚体,1ml叔戊醇,在160℃下搅拌反应16小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到四氢萘啶并四氢喹唑啉化合物2a;
所得产物2a的氢谱图及碳谱图分别如图5和图6所示,结构表征数据如下:
1hnmr(400mhz,cdcl3):δ7.85–7.78(m,2h),7.47–7.41(m,4h),6.76(d,j=6.2hz,2h),6.03(s,1h),4.48(s,1h),3.34–3.24(m,2h),2.84–2.70(m,4h),2.16(s,3h),2.10–2.02(m,2h),1.93–1.82(m,2h);
13cnmr(101mhz,cdcl3):δ159.69,157.72,139.84,139.19,137.85,129.80,129.49,128.59,128.39,125.84,125.05,124.91,122.55,119.84,114.67,94.24,52.07,42.33,27.57,25.39,24.84,22.03,20.51;
ir(kbr):3408,3056,2939,2844,2211,1711,1602,1507,1435,1279,1189,918,745,700cm-1;
yellowsolid,m.p:197-199℃;
hrms(esi):calcd.forc25h24n4[m+h]+:381.2074;found:381.2080。
根据以上数据推断所得产物2a的结构如下式所示:
(2)在反应器中,加入0.1毫摩尔2a与0.1毫摩尔苯甲醛,加入1ml冰醋酸,在100℃下搅拌反应2小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到四氢萘啶并四氢喹唑啉类化合物2b。
所得产物2b的氢谱图及碳谱图分别如图7和图8所示,结构表征数据如下:
1hnmr(400mhz,cdcl3):δ7.96(dd,j=7.8,1.5hz,2h),7.54–7.46(m,4h),7.44(s,1h),7.36(d,j=6.5hz,2h),7.32–7.27(m,3h),6.76(s,1h),6.70(s,1h),4.51(t,j=5.4hz,1h),3.47–3.30(m,2h),2.92–2.65(m,4h),2.31–2.25(m,2h),2.23(s,3h),2.09–1.96(m,2h);
13cnmr(101mhz,cdcl3):δ157.62,154.56,139.09,138.98,137.23,136.99,128.47,127.70,127.65,127.63,127.35,126.78,126.00,123.91,121.74,120.62,119.83,118.75,115.51,93.64,67.89,49.14,47.23,26.56,23.93,23.05,20.73,19.58;
ir(kbr):3047,2926,2327,1733,1701,1592,1493,1270,1016,854,807,742cm-1;
brownishsolid,m.p:240-242℃;
hrms(esi):calcd.forc32h28n4na[m+h]+:491.2206;found:491.2207。
根据以上数据推断所得产物2b的结构如下式所示
实施例3
(1)在反应器中,加入0.3毫摩尔四氢喹啉和0.3毫摩尔2-苯基3-腈基1,8萘啶,0.05毫摩尔二氯(五甲基环戊二烯基)合铱(iii)二聚体,1ml对二甲苯,在160℃下搅拌反应12小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到1a;
(2)在反应器中,加入0.1毫摩尔1a与0.15毫摩尔4-溴苯甲醛,加入1ml甲酸,在50℃下搅拌反应10小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到四氢萘啶并四氢喹唑啉化合物3b。
所得产物3b的氢谱及碳谱分别如图9和图10所示,结构表征数据如下:
1hnmr(400mhz,cdcl3):δ7.97(dd,j=7.7,1.6hz,2h),7.55–7.51(m,3h),7.49(s,1h),7.46(d,j=8.0hz,3h),7.29(d,j=3.7hz,2h),6.96(d,j=7.4hz,1h),6.92(d,j=7.6hz,1h),6.65(t,j=7.5hz,1h),4.53(t,j=5.2hz,1h),3.52–3.43(m,1h),3.42–3.33(m,1h),2.96–2.75(m,4h),2.38–2.23(m,2h),2.14–1.99(m,2h);
13cnmr(101mhz,cdcl3):δ158.67,155.42,140.31,140.08,138.99,138.13,131.85,129.60,128.83,128.64,128.45,128.17,122.32,121.90,121.74,120.73,119.57,116.65,116.17,95.17,68.65,50.03,48.33,27.61,24.77,23.89,21.54;
ir(kbr):2923,2852,1599,1538,1459,1440,1311,1271,1147,1010,811,743,698cm-1;
brownishsolid,m.p:111-113℃;
hrms(esi):calcd.forc31h25n4na[m+h]+:555.1155;found:555.1150。
根据以上数据推断所得产物3b的结构如下式所示
实施例4
(1)在反应器中,加入0.3毫摩尔四氢喹啉和0.45毫摩尔2-苯基3-腈基1,8萘啶,0.01毫摩尔二氯(五甲基环戊二烯基)合铱(iii)二聚体,1ml二氧六环,在100℃下搅拌反应18小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到1a;
(2)在反应器中,加入0.1毫摩尔1a与0.15毫摩尔4-甲氧基苯甲醛,加入1ml冰乙酸,在80℃下搅拌反应3小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到四氢萘啶并四氢喹唑啉化合物4b。
所得产物4b的氢谱及碳谱分别如图11和图12所示,结构表征数据如下:
1hnmr(400mhz,cdcl3):δ7.99–7.94(m,2h),7.54–7.47(m,3h),7.44(s,2h),7.28(d,j=8.6hz,2h),6.92(d,j=7.4hz,1h),6.89(d,j=7.6hz,1h),6.83(d,j=8.7hz,2h),6.60(t,j=7.5hz,1h),4.54(t,j=5.2hz,1h),3.77(s,3h),3.45–3.35(m,2h),2.89–2.68(m,4h),2.31–2.24(m,2h),2.10–1.97(m,2h);
13cnmr(101mhz,cdcl3):δ159.28,158.65,155.52,140.44,140.11,138.26,131.85,129.50,128.67,128.38,128.17,128.03,122.23,121.50,120.77,119.74,116.55,115.76,114.04,94.67,68.64,55.27,49.89,48.20,27.67,24.74,23.97,21.53;
ir(kbr):3049,2928,2839,1726,1600,1538,1492,1311,1246,1034,742,698cm-1;
brownishoilliquid;
hrms(esi):calcd.forc32h28n4ona[m+h]+:507.2155;found:507.2158。
根据以上数据推断所得产物4b的结构如下式所示
实施例5
(1)在反应器中,加入0.5毫摩尔四氢喹啉和0.3毫摩尔2-苯基3-腈基1,8萘啶,0.01毫摩尔1,5-环辛二烯氯化铱二聚体,1ml对二甲苯,在130℃下搅拌反应20小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到1a;
(2)在反应器中,加入0.1毫摩尔1a与0.2毫摩尔乙醛,加入1.5ml冰乙酸,在60℃下搅拌反应8小时,反应结束后冷却至室温,稀释反应液,过滤,减压旋蒸除去溶剂即可得到粗产物,粗产物经柱层析提纯得到四氢萘啶并四氢喹唑啉化合物5b。
所得产物5b的氢谱及碳谱分别如图13和图14所示,结构表征数据如下:
1hnmr(400mhz,cdcl3):δ7.92(dd,j=7.8,1.5hz,2h),7.56–7.46(m,3h),7.44(s,1h),6.97(d,j=7.6hz,1h),6.91(d,j=7.2hz,1h),6.68(t,j=7.5hz,1h),6.33(q,j=6.2hz,1h),4.83(dd,j=10.0,3.1hz,1h),3.37–3.25(m,1h),3.22–3.12(m,1h),3.02–2.87(m,2h),2.85–2.71(m,2h),2.56–2.44(m,1h),2.15–1.98(m,3h),1.32(d,j=6.2hz,3h);
13cnmr(101mhz,cdcl3):δ158.71,154.83,139.86,139.54,138.35,129.45,128.67,128.33,127.90,123.33,123.25,121.51,119.89,116.92,115.86,93.98,62.62,51.82,46.91,28.85,27.48,25.78,21.62,13.16;
ir(kbr):2926,2851,2212,1601,1538,1462,1319,1205,1085,747,698cm-1;
brownishoilliquid;
hrms(esi):calcd.forc26h25n4[m+h]+:393.2074;found:393.2073。
根据以上数据推断所得产物5b的结构如下式所示
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!