用于制造2,3,3,3-四氟丙烯的改进的方法与流程
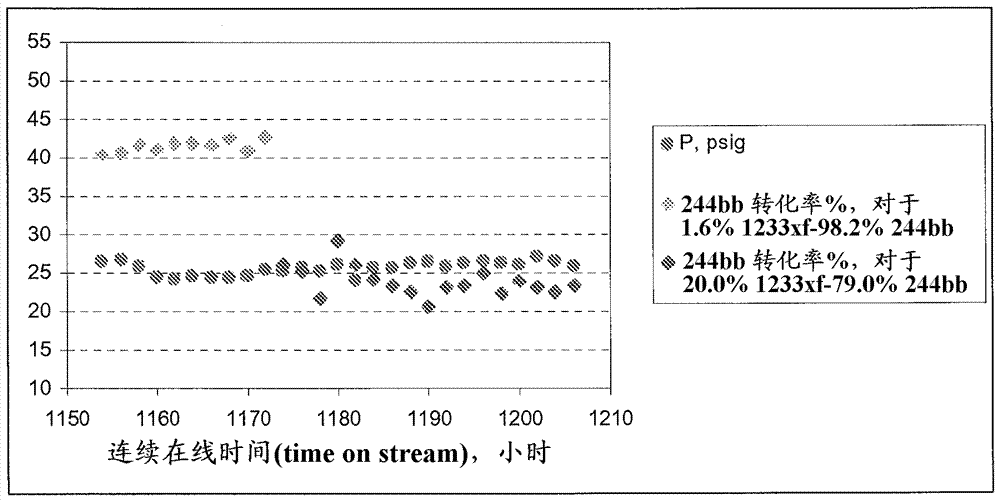
相关申请本申请要求2012年2月10日提交到美国专利局的ussn61/597,672的优先权,其内容通过引用而结合。发明领域本发明涉及一种用于制备氟化的有机化合物的方法,更特别是,本发明涉及一种用于制备氟化烯烃的方法,甚至更特别是,本发明涉及一种用于制备2,3,3,3-四氟丙烯(hfo-1234yf)的方法。发明背景某些氢氟烯烃(hfo)例如四氟丙烯(包括2,3,3,3-四氟丙烯(hfo-1234yf))已知为有效的致冷剂、灭火剂、传热介质、推进剂、起泡剂、发泡剂、气态电介质、消毒剂载体、聚合介质、颗粒去除流体、载体流体、缓冲研磨剂、置换干燥剂和动力循环工作流体。与大多数氯氟烃(cfc)和氢氯氟烃(hcfc)不同,大多数hfo对臭氧层不造成威胁。hfo-1234yf已显示为具有低毒性的低全球变暖化合物,因此,可满足在移动空气调节装置中对于致冷剂的日益严格的要求。因此,含有hfo-1234yf的组合物是正开发用于许多前述应用的材料中的领导者。制备hfo的若干方法为已知的。例如,美国专利号4,900,874(ihara等人)描述了一种通过使氢气与氟化的醇接触制备含氟烯烃的方法。虽然这似乎是相对高收率的过程,但在高温下工业规模处理氢气有潜在危险。另外,工业生产氢气(例如建造原地氢设备)的成本在经济上昂贵。美国专利号2,931,840(marquis)描述了一种通过氯代甲烷和四氟乙烯或一氯二氟甲烷的热解,制备含氟烯烃的方法。该方法为相对低收率的过程,并且非常大百分比的有机原料转化为不需要的和/或不重要的副产物,包括相当大量的炭黑,其倾向于使在过程中使用的催化剂失活。已描述由三氟乙酰基丙酮和四氟化硫制备hfo-1234yf(参见banks等人,journaloffluorinechemistry,第82卷,第2期,第171-174页(1997))。另外,美国专利号5,162,594(krespan)公开了一种其中四氟乙烯与另一种氟化的乙烯在液相中反应以生产聚氟烯烃产物的过程。虽然上述过程和其它过程总体上用于生产氟化烯烃,特别是生产氟化丙烯,但认识到,仍需要总体上生产氟化烯烃,特别是生产氟化丙烯(例如hfo-1234yf)的更经济有效的手段。本发明尤其满足该需求。概述本发明的一方面提供一种用于形成2,3,3,3-四氟丙烯(hfo-1234yf)的方法,所述方法包括提供具有相对低浓度的2-氯-3,3,3-三氟丙烯(hcfo-1233xf)的脱氢氯化原料,和在金属合金存在下使所述原料脱氢氯化。不希望束缚于任何具体的操作理论,本发明的某些方面基于以下观察和理解,在某些脱氢氯化原料(例如2-氯-1,1,1,2-四氟丙烷(hcfc-244bb))的某些脱氢氯化反应以生产2,3,3,3-四氟丙烯(hfo-1234yf)期间,在反应原料(例如hcfc-244bb原料)中存在2-氯-3,3,3-三氟丙烯(hcfo-1233xf)可导致显著降低的hcfc-244bb到hfo-1234yf的转化并且增加形成3,3,3-三氟丙炔(cf3cch),这是2-氯-3,3,3-三氟丙烯(hcfo-1233xf)的脱氢氯化产物。此外,当经历脱氢氯化时,2-氯-3,3,3-三氟丙烯共存于原料中,可导致形成低聚物,这可生产焦油。该结果不仅从期望产物的降低收率的观点来看是不利的,其另外还不利,因为3,3,3-三氟丙炔(cf3cch)副产物为易燃气体和有毒,因此是不期望的。因此,本发明部分涉及通过基本上降低和/或基本上消除在反应器进料中(尤其是在hcfc-244bb进料流中)hcfo-1233xf的存在,改进在某些脱氢氯化反应中脱氢氯化原料(例如hcfc-244bb)的转化和降低不需要的反应副产物(例如cf3cch)的量的方法。在一方面,本发明涉及一种用于制备氟烯烃的原料,尤其是在脱氢氯化反应中,这在下文中描述。在一个实施方案中,在金属合金存在下发生脱氢氯化反应,在另一实施方案中,使用镍金属合金。在另一实施方案中,不使用基于氯化铯的催化剂,进行脱氢氯化反应。在其它实施方案中,在包含金属合金的反应器内发生反应。在本发明的另一方面,在包含镍金属合金的反应器中进行反应,在另一实施方案中,反应器包含奥氏体镍基材料,例如inconel625。在这些实施方案的其它方面,使用上述实施方案的任一种进行反应,例如包含奥氏体镍基材料例如inconel625的反应器,并且基本不存在加入的基于cscl的催化材料或颗粒,在某些实施方案中,基本不存在任何加入的催化材料或颗粒。对于所有这些实施方案,通向反应器的原料或进料流具有低于约20%重量的2-氯-3,3,3-三氟丙烯。在另一实施方案中,原料或进料流含有低于约8.0%重量的hcfo-1233xf(如下文定义的),而在其它实施方案中,其含有低于约2%重量的hcfo-1233xf,在一些其它实施方案中,低于约1%重量的hcfo-1233xf。在另一实施方案中,进料流或原料包含基本上不含hcfo-1233xf的2-氯-1,1,1,2-四氟丙烷。在本发明的一方面,原料或有机进料流以主要比例包含hcfc-244bb,其为基本上纯的或足够纯,使得通过任何设备不能检测到在组合物中存在hcfo-1233xf。本文使用的术语“包含”、“包括”、“具有”或它们的任何其它变体旨在涵盖非排他的内含物。例如,包含要素列举的过程、方法、制品或设备不必仅局限于那些要素,而是可包括未明确列举的或该过程、方法、制品或设备固有的其它要素。此外,除非明确说明相反的情况,否则“或”指包括性的“或”而不是排除性的“或”。例如,以下任一种情况满足条件a或b:a为真(或存在)并且b为假(或不存在),a为假(或不存在)并且b为真(或存在),以及a和b二者均为真(或存在)。另外,使用“一个”用于描述本文描述的要素和组分。这样做仅为了方便和给出本发明范围的概要含义。该描述应理解为包括一个或至少一个,并且单数还包括复数,除非明显其另有所指。除非另外定义,否则本文使用的所有技术和科学术语具有与本发明所属领域技术人员通常理解相同的含义。在冲突的情况下,以本说明书(包括定义)为准。虽然与本文描述的那些类似或等同的方法和材料可用于实践或测试本发明的实施方案,但以下描述合适的方法和材料。此外,材料、方法和实施例仅为说明性的并且不旨在限制。当按照范围、优选范围或上优选值和/或下优选值的列举给出量、浓度或其它值或参数时,应理解为明确公开了由任何上限范围或优选值和任何下限范围或优选值的任何配对形成的所有范围,无论该范围是否单独公开。当本文引用数值的范围时,除非另外说明,否则该范围旨在包括其端点以及在该范围内的所有整数和分数。关于在进料流或原料中hcfo-1233xf的量,本文使用的术语“重量%”指基于通向反应器的进料中的脱氢氯化反应物和1233xf的总有机部分,hcfo-1233xf的重量百分数。本文使用的术语“基本不含”意味着指低于约0.3%重量。在本发明的实施方案中,尤其是包含脱氢氯化反应的那些实施方案,hcfo-1233xf以低于约20重量%的量存在于反应材料中。在另一实施方案中,hcfo-1233xf以低于15重量%存在,而在另一方面,其以低于约10重量%存在。在本发明的另一方面,其以低于约8重量%存在。在某些实施方案中,hcfo-1233xf以低于约5重量%的量存在于反应材料中,在另一实施方案中,低于约2.5重量%,在另一实施方案中,低于约1.0重量%。在某些实施方案中,进料材料基本上不含hcfo-1233xf。在另一方面,本发明涉及一种用于降低在脱氢氯化反应进料(例如hcfc-244bb原料)内hcfo-1233xf的水平的方法,通过提供含有hcfo-1233xf的包含脱氢氯化反应进料材料(例如,hcfc-244bb)的组合物,和降低hcfo-1233xf的水平,使得存在的hcfo-1233xf的量占低于约-20%重量,在一个实施方案中,低于15重量%,在另一实施方案中,低于约10重量%,在另一实施方案中,低于约8重量%,还低于约5重量%、低于约2.5%,在另一实施方案中,低于约1重量%。在本发明的另一方面,脱氢氯化反应进料材料基本上不含hcfo-1233xf。本文描述的脱氢氯化反应可使用本文描述的原料或进料流在液相或蒸气相中进行。在一个实施方案中,脱氢氯化反应不使用实质量的催化剂或使用包含奥氏体镍基材料的催化剂。虽然降低hcfo-1233xf的水平的方法可包括本文提供的任何一种方法或多种方法的组合,但在某些方面,所述方法包括以下之一或任何组合:(i)将组合物蒸馏,以将hcfo-1233xf与脱氢氯化反应进料(例如hcfc-244bb)分离。在某些实施方案中,蒸馏方法可包括萃取蒸馏,其中使用足以改变2-氯-3,3,3-三氟丙烯和/或2-氯-1,1,1,2-四氟丙烷的挥发性的体积的萃取剂或溶剂,萃取hcfo-1233xf或hcfc-244bb。随后将组合物蒸馏,以提供2-氯-1,1,1,2-四氟丙烷作为馏出物。这样的剂或溶剂可包括但不限于反式-hcfo-1233zd、顺式-hcfo-1233zd、hcfo-1223异构体和hcfo-1232异构体和它们的组合;(ii)用过量的hf处理起始组合物,以在催化剂存在下,在后催化反应器中,将未反应的hcfo-1233xf转化为hcfc-244bb。在某些方面,在约70℃-约120℃温度和/或约50psig-约120psig压力下,用过量的hf处理组合物。(iii)使用共沸蒸馏处理起始组合物。在某些实施方案中,这样的处理包括以有效量向组合物中加入第三组分,以与hcfo-1233xf、脱氢氯化反应进料(例如hcfc-244bb)形成三元共沸物,和/或与hcfo-1233xf和脱氢氯化反应进料(例如hcfc-244bb)中的一种或二者形成二元共沸物。随后使用标准分离技术(例如但不限于蒸馏)从组合物除去共沸物;(iv)使所述起始组合物光氯化,以使hcfo-1233xf饱和并形成1,2,2-三氯-3,3,3-三氟丙烷(hcfc-233ab)。在某些实施方案中,光氯化处理包括在至少一种氯化剂存在下,使组合物暴露于辐射(例如,紫外光)。随后使用标准方法(例如但不限于蒸馏)从组合物除去hcfc-233ab;(v)将起始组合物冷却至其中2-氯-1,1,1,2-四氟丙烷和2-氯-3,3,3-三氟丙烯中的一个形成固体而另一个保持为液体的温度。在某些实施方案中,将组合物冷却至其中2-氯-1,1,1,2-四氟丙烷形成固体而2-氯-3,3,3-三氟丙烯为液体的温度。随后除去2-氯-1,1,1,2-四氟丙烷。在某些实施方案中,将组合物冷却至约-75℃至-85℃的温度。(vi)使所述起始组合物通过选自活性炭和沸石和/或分子筛的固体吸附剂的固定床,其中对沸石和/或分子筛中的孔选定尺寸以从2-氯-3,3,3-三氟丙烯过滤2-氯-1,1,1,2-四氟丙烷。在某些方面,孔为约3å-约10å;(vii)通过层析法将hcfo-1233xf与脱氢氯化反应进料(例如hcfc-244bb)分离,其中使hcfo-1233xf和hcfc-244bb的混合物进料通过含有固定基材的柱;和(viii)使所述起始组合物与液体氢氟酸混合,以形成包含2-氯-1,1,1,2-四氟丙烷、2-氯-3,3,3-三氟丙烯和hf的混合物,所述hf以低于起始组合物的约10%重量存在,且在再沸器中蒸馏所得到的混合物,收集产物,该产物基本上不含hcfo-1233xf,并且2-氯-1,1,1,2-四氟丙烷比起在所得到的起始混合物中更纯。在其它实施方案中,本发明涉及一种用于制备基本上不含3,3,3-三氟丙炔的hfo-1234yf的方法,通过提供包括基本上不含hcfo-1233xf的hcfc-244bb的起始组合物,使所述起始组合物与脱氢氯化催化剂接触,以生产包含基本上不含3,3,3-三氟丙炔的hfo-1234yf的最终的组合物。在某些实施方案中,脱氢氯化催化剂选自(i)一种或多种金属卤化物,(ii)一种或多种卤化金属氧化物,(iii)一种或多种零价金属/金属合金,(iv)奥氏体镍基材料(例如inconel625)和(v)这些中两个或更多个的组合。在某些实施方案中,脱氢氯化催化剂主要不包含,甚至更优选不含任何实质量的基于氯化铯(cscl)的催化剂以及基于氟化镁(mgf2)的催化剂。在另一实施方案中,本发明涉及制备基本上不含3,3,3-三氟丙炔的2,3,3,3-四氟丙烯的方法,所述方法包括:(i)提供包含基本上不含2-氯-3,3,3-三氟丙烯的2-氯-1,1,1,2-四氟丙烷的起始组合物;和(ii)使所述起始组合物与脱氢氯化催化剂接触,以生产包含2,3,3,3-四氟丙烯的最终组合物,所述最终的组合物基本上不含3,3,3-三氟丙炔。在一种实施方案中,制备2,3,3,3-四氟丙烯的方法另外包括测量存在于最终组合物中的3,3,3-三氟丙炔的量。在甚至其它方面,本发明涉及一种用于制备2,3,3,3-四氟丙烯的方法,所述方法通过:(i)提供包括式i、ii或iii的化合物的起始组合物:cx2=ccl-ch2x (i);cx3-ccl=ch2 (ii);或cx3-chcl-ch2x (iii)其中x独立地选自f、cl、br和i,条件是至少一个x不是氟;(ii)使所述起始组合物与第一氟化剂接触,以生产包括hcfo-1233xf和第一含氯副产物的第一中间组合物;(iii)使所述第一中间组合物与第二氟化剂接触,以生产包括hcfc-244bb和hcfo-1233xf的第二中间组合物;(iv)降低在所述第二中间组合物中的hcfo-1233xf的水平,以生产包括基本上不含hcfo-1233xf的hcfc-244bb的第三中间组合物;和(v)使至少一部分hcfc-244bb脱氢氯化,以生产包括基本上不含3,3,3-三氟丙烯的hfo-1234yf的反应产物。基于本文提供的公开内容,本发明的另外的实施方案和优点对于本领域技术人员显而易见。附图简述图1说明在由奥氏体镍基材料(特别是inconel625)组成的反应容器中,hcfo-1233xf浓度对hcfc-244bb到hfo-1234yf的转化的效果,在气相反应中压力相对时间。优选实施方案的详述根据一个实施方案,本发明涉及一种使用包含基本上不含hcfo-1233xf的hcfc-244b的起始或中间材料,用于制备hfo-1234yf的制造方法。本发明人意外地发现存在hcfo-1233xf降低hcfc-244bb到hfo-1234yf的转化,hcfc-244bb到hfo-1234yf的转化导致更大的再循环、提高的设备成本和更高的操作成本。此外,存在hcfo-1233xf增加形成3,3,3-三氟丙炔(cf3cch),其为易燃气体因此为不期望的。其从最终产物(即hfo-1234yf)的去除将导致额外的成本并且还可能导致收率损失。例如,使用蒸馏使cf3cch水平从50-100ppm降至5-12ppm引起接近10%的hfo-1234yf损失。因此,本发明提供从反应器进料除去hcfo-1233xf的方法,以改进hfo-1234yf转化过程的总体效率。在某些方面,制备hfo-1234yf通常包括如下的至少三个反应步骤:(i)在装载固体催化剂的蒸气相反应器中,(cx2=ccl-ch2x或cx3-ccl=ch2或cx3-chcl-ch2x)+hf→2-氯-3,3,3-三氟丙烯(hcfo-1233xf)+hcl;(ii)在装载液体氢氟化催化剂的液相反应器中,2-氯-3,3,3-三氟丙烯(hcfo-1233xf)+hf→2-氯-1,1,1,2-四氟丙烷(hcfc-244bb);和(iii)在蒸气相反应器中,2-氯-1,1,1,2-四氟丙烷(hcfc-244bb)→2,3,3,3-四氟丙烯(hfo-1234yf)。其中x独立地选自f、cl、br和i,条件是至少一个x不是氟。通常来说,第一反应步骤的原料可用一种或多种式i、ii和/或iii的氯化化合物表示:cx2=ccl-ch2x (式i)cx3-ccl=ch2 (式ii)cx3-chcl-ch2x (式iii)其中x独立地选自f、cl、br和i,条件是至少一个x不是氟。在某些实施方案中,这些化合物含有至少一个氯,大多数x为氯,或者所有的x为氯。在第一步骤中,这些原料(在某些实施方案中,其包括1,1,2,3-四氯丙烯(1230xa)和/或1,1,1,2,3-五氯丙烷(hcc-240db))与无水hf在第一蒸气相反应器(氟化反应器)中反应,以生产至少hcfo-1233xf(2-氯-3,3,3-三氟丙烯)和hcl的混合物。反应可在约200-400℃温度和约0-200psig压力下进行。离开蒸气相反应器的流出物流可任选包含另外的组分,例如未反应的hf、重质中间体、hcfc-244bb、hfc-245cb(1,1,1,2,2-五氟丙烷)等。该反应可在适合蒸气相氟化反应的任何反应器中进行。反应器可由耐氟化氢的腐蚀效果的材料和催化剂(例如hastalloy、inconel、monel)构成。在蒸气相过程的情况下,反应器填充有蒸气相氟化催化剂。本领域已知的任何氟化催化剂可用于该过程。合适的催化剂包括但不限于铬、铝、钴、锰、镍和铁的氧化物、氢氧化物、卤化物、卤氧化物、其无机盐和它们的混合物,其中的任一种可任选被卤化。适用于本发明的催化剂的组合非排他地包括cr2o3、fecl3/c、cr2o3/al2o3、cr2o3/alf3、cr2o3/碳、cocl2/cr2o3/al2o3、nicl2/cr2o3/al2o3、cocl2/alf3、nicl2/alf3和它们的混合物。氧化铬/氧化铝催化剂描述于美国专利号5,155,082,其内容通过引用结合到本文中。优选铬(iii)氧化物,例如结晶氧化铬或无定形氧化铬,最优选无定形氧化铬。氧化铬(cr2o3)为市售可得的材料,其可按多种粒径购买。优选具有至少98%纯度的氟化催化剂。氟化催化剂过量存在,但是有至少足以驱动反应的量。反应的该第一步骤不必局限于如上所述的蒸气相反应,而是也可使用液相反应或液相和蒸气相的组合实施,例如在美国公布专利申请号20070197842中公开的,其内容通过引用结合到本文中。还预期反应可分批进行、连续进行或这些的组合。对于其中反应包含液相反应的实施方案,反应可为催化或非催化的。可采用路易斯酸催化剂,例如金属-卤化物催化剂,包括锑卤化物、锡卤化物、铊卤化物、铁卤化物和这些的两个或更多个的组合。在某些实施方案中,采用金属氯化物和金属氟化物,包括但不限于sbcl5、sbcl3、sbf5、sncl4、ticl4、fecl3和这些的两个或更多个的组合。来自反应器的流出物可任选经处理,以实现期望程度的分离和/或其它处理。通过非限制性实例,产物流出物可含有一种或多种杂质,例如,hcl、未转化的反应物和/或其它副产物。这些产物可使用已知的或本文另外讨论的标准方法除去。hcl例如可通过常规的蒸馏回收,或者使用水或碱洗涤器,如以下更详细讨论的,将未反应的起始试剂分离和再循环。在用于形成2,3,3,3-四氟丙烯的过程的第二步骤中,hcfo-1233xf转化为hcfc-244bb。在一个实施方案中,该步骤可在液相反应器中在液相中实施,该液相反应器可为tfe或pfa衬里的。这样的过程可在约70-120℃温度范围和约50-120psig下实施。任何液相氟化催化剂可用于本发明。非穷举的列举包括路易斯酸、过渡金属卤化物、过渡金属氧化物、ivb族金属卤化物、vb族金属卤化物或它们的组合。液相氟化催化剂的非排他实例为卤化锑、卤化锡、卤化钽、卤化钛、卤化铌、卤化钼、卤化铁、氟化的卤化铬、氟化的氧化铬,或它们的组合。液相氟化催化剂的具体非排他实例为sbcl5、sbcl3、sbf5、sncl4、tacl5、ticl4、nbcl5、mocl6、fecl3、sbcl5的氟化物类、sbcl3的氟化物类、sncl4的氟化物类、tacl5的氟化物类、ticl4的氟化物类、nbcl5的氟化物类、mocl6的氟化物类、fecl3的氟化物类,或它们的组合。最优选五氯化锑。如果变得失活,这些催化剂可容易通过本领域已知的任何手段再生。使催化剂再生的一种合适的方法涉及使氯流流动通催化剂。例如,对于每磅液相氟化催化剂,可向液相反应中加入约0.002-约0.2lb/小时的氯。这可例如在约65℃-约100℃的温度下进行约1-约2小时或连续进行。反应的该第二步骤不必局限于液相反应,并且也可使用蒸气相反应或液相和蒸气相的组合实施,例如在美国公布专利申请号20070197842中公开的,其内容通过引用结合到本文中。为此,将含有hcfo-1233xf的进料流预热至约50℃-约400℃的温度,并且与催化剂和氟化剂接触。催化剂可包括用于这样的反应的标准蒸气相剂,并且氟化剂可包括本领域通常已知的那些,例如但不限于氟化氢。在hfo-1234yf生产的第三步骤中,将hcfc-244bb进料至第二蒸气相反应器(脱氢氯化反应器)经脱氢氯化,以制备期望的产物2,3,3,3-四氟丙烯(hfo-1234yf)。该反应器可为非催化的,或者其可含有可使hcfc-244bb催化脱氢氯化的催化剂,以制备hfo-1234yf。催化剂(如果存在)可为金属卤化物、卤化金属氧化物、中性(或零氧化态)金属或金属合金或整体式或担载式活性炭。金属卤化物或金属氧化物催化剂可包括但不限于一价、二价和三价金属卤化物、氧化物和它们的混合物/组合,更优选一价和二价金属卤化物和它们的混合物/组合。组分金属包括但不限于cr3+、fe3+、mg2+、ca2+、ni2+、zn2+、pd2+、li+、na+、k+和cs+。组分卤素包括但不限于f-、cl-、br-和i-。可用的一价或二价金属卤化物的实例包括但不限于lif、naf、kf、csf、mgf2、caf2、licl、nacl、kcl和cscl。卤化处理可包括任何现有技术已知的那些,特别是采用hf、f2、hcl、cl2、hbr、br2、hi和i2作为卤化源的那些。当为中性(即,零价)时,使用金属、金属合金和它们的混合物。可用的金属包括但不限于pd、pt、rh、fe、co、ni、cu、mo、cr、mn和作为合金或混合物的前述物质的组合。催化剂可为担载的或非担载的。金属合金的可用实例包括但不限于ss316、monel400、incoloy825、alloy20、hastelloy、inconel600和inconel625。在本发明的一方面,催化剂包括活性炭、不锈钢(例如,ss316)、奥氏体镍基合金(例如,inconel625)、镍,在某些实施方案中,包括氟化的10%cscl/mgo。合适的反应温度为约300-550℃,合适的反应压力可为约0-150psig。反应器流出物可进料至碱洗涤器或蒸馏塔,以除去hcl的副产物,以生产不含酸的有机产物,使用本领域已知的纯化技术中的一种或任何组合,其任选可经历进一步纯化。反应可在约200℃-约800℃,约300℃-约600℃,或约400℃-约500℃温度范围下进行。合适的反应器压力在约0psig-约200psig,约10psig-约100psig,或约20-约70psig范围。总的来说,来自脱氢氯化反应器的流出物可经处理,以实现期望程度的分离和/或其它处理。除了生产的hfo-1234yf以外,流出物通常还含有hcl、未转化的hcfc-244bb和hcfo-1233xf(其主要由hcfo-1233xf氢氟化的先前步骤带入)。任选,随后从脱氢氯化反应的产物回收hcl。通过常规的蒸馏进行hcl回收,其中将hcl从馏出物除去。或者,通过使用水或碱洗涤器可回收或除去hcl。当使用水萃取器时,除去hcl,为含水溶液。当使用碱溶液时,从系统除去hcl,为含水溶液中的氯化物盐。在回收或去除hcl后,可将有机流送至蒸馏塔用于分离。从柱的塔顶物收集的hfo-1234yf可送至另一个柱,用于进一步纯化,同时可将在再沸器累积的一部分hcfo-1233xf和hcfc-244bb的混合物送回脱氢氯化反应器,用于再循环hcfc-244bb,将剩余的送到hcfo-1233xf氢氟化反应器,用于再循环hcfo-1233xf。本发明人意外地观察到,在脱氢氯化反应物(例如hcfc-244bb)脱氢氯化以形成hfo-1234yf期间,在原料中存在hcfo-1233xf降低反应物(例如hcfc-244bb)到hfo-1234yf的转化,并且增加形成3,3,3-三氟丙炔(一种不期望的副产物)。较低转化的结果包括试剂再循环的较高体积,每一个反应使用较大的设备,和总体较高的操作成本。形成三氟丙炔的结果包括在其去除中导致的额外成本和1234yf的收率损失。本发明提供一种对至少这些问题的解决方案,通过降低在进料流中hcfo-1233xf的含量,从而改进hfo-1234yf生产效率。为此,在第三反应步骤之前,在一个实施方案中,原料组合物包含hcfc-244bb,在另一实施方案中,基本上由hcfc-244bb组成,并且优选首先经纯化,以形成含有低于约20%重量的hcfo-1233xf的原料。在另一实施方案中,进料流含有低于约8.0重量%的hcfo-1233xf。在再一个实施方案中,进料流基本上不含hcfo-1233xf。在一方面,以低于约20.0重量%,低于约15.0重量%,低于约10.0重量%,低于约8.0重量%,低于约5.0重量%,低于约2.5重量%,低于约1.0重量%的量,在hcfc-244bb的纯化原料中提供hcfo-1233xf。在另一实施方案中,用于脱氢氯化步骤的进料流基本上不含hcfo-1233xf。在其它实施方案中,用于脱氢氯化反应的原料包含2-氯-1,1,1,2-四氟丙烷和2-氯-3,3,3-三氟丙烯,其中2-氯-3,3,3-三氯丙烯的浓度为20%或19%或18%或17%或16%或15%或14%或13%或12%或11%或19%或9%或8%或7%或6%或5%或4%或3%或2%或1%或0.9%或0.8%或0.7%或0.6%或0.5%或0.4%或0.3%,以重量计。在另一实施方案中,用于脱氢氯化的原料包含基本上不含2-氯-3,3,3-三氯丙烯的2-氯-1,1,1,2-四氟丙烷。在本发明的一方面,原料溶液包含至少约80重量%的2-氯-1,1,1,2-四氟丙烷,在另一实施方案中,有至少约85重量%的2-氯-1,1,1,2-四氟丙烷,在另一实施方案中,有至少约90重量%的2-氯-1,1,1,2-四氟丙烷,而在另一实施方案中,其包含至少约92重量%的2-氯-1,1,1,2-四氟丙烷,而在再一实施方案中,至少约95重量%的2-氯-1,1,1,2-四氟丙烷。在又一实施方案中,原料包含至少约97.5重量%的2-氯-1,1,1,2-四氟丙烷,在另一实施方案中,至少约99重量%的2-氯-1,1,1,2-四氟丙烷,在又一实施方案中,至少约99.7重量%的2-氯-1,1,1,2-四氟丙烷。在又一实施方案中,原料包含以下实施方案:80重量%,81重量%,82重量%,83重量%,84重量%,85重量%,86重量%,87重量%,88重量%,89重量%,90重量%,91重量%,92重量%,93重量%94重量%,95重量%96重量%,97重量%,98重量%99重量%,99.1重量%,99.2重量%,99.3重量%,99.4重量%,99.5重量%,99.6重量%,99.7重量%或更高的2-氯-1,1,1,2-四氟丙烷。在某些方面,在一个实施方案中,这样的组合物将hcfc-244bb到hfo-1234yf的转化率提高至最少20%或更高,在另一实施方案中,25%或更高,或者在再一实施方案中,30%或更高,在本发明的另一方面,35%或更高。此外,在一个实施方案中,在脱氢氯化反应中cf3cch的形成降低至至少1000ppm或更低,在另一实施方案中,500ppm或更低,在第三实施方案中,400ppm或更低,在另一实施方案中,300ppm或更低,或者在又一实施方案中,200ppm或更低。通过用形成的产物(hfo-1234yf)的摩尔数除以消耗的反应物的摩尔数,或者另外使用本领域已知的标准方法,可计算转化率和/或选择性。本领域已知的任何技术可用于纯化脱氢氯化反应进料(例如hcfc-244bb),或者另外除去hcfo-1233xf。提供以下分离方法作为这样的纯化方法的实施方案,但是不必局限于本发明。这样的实施方案可单独实施,以任何组合实施,和/或使用一种或多种本领域另外已知的备选分离方法。在一个实施方案中,可使用蒸馏和/或萃取蒸馏将hcfo-1233xf与hcfc-244bb流分离。如本领域已知的,蒸馏为基于两种化合物的挥发性的差异,将hcfc-244bb与hcfo-1233xf分离。在本发明中,基于成分的沸腾温度、环境压力等,可使用标准方法实施这样的技术。萃取蒸馏指使用萃取剂或溶剂将hcfo-1233xf或hcfc-244bb中的一种与另一种分离。如在美国专利号7,803,283中描述的,其内容通过引用结合到本文中,本领域已知,当以某些组分量共同提供时,hcfo-1233xf和hcfc-244bb呈现共沸性质。为了将一种组分与另一种分离,可向这样的溶液中加入萃取剂。本文使用的术语“萃取剂”指一种化合物或多种化合物的混合物,其改变hcfo-1233xf和hcfc-244bb中的一个或二者的挥发性,以允许使用标准手段(例如,蒸馏)分离这些化合物。优选,但不是排他地,这样的剂具有低挥发性,并且不与hcfo-1233xf和hcfc-244bb中的任一个或二者形成第二共沸物或类似共沸物的组合物。通过非限制性实例,在一个实施方案中,以高浓度向hcfo-1233xf和hcfc-244bb的混合物中加入萃取剂。将组合物蒸馏,随后从混合物取出hcfo-1233xf或hcfc-244bb中的任一个作为馏出物。将包括剩余组分和萃取剂的底部产物分离,任选纯化至所需的或期望的程度。在本发明的某些实施方案中,萃取剂可为促进hcfc-244bb作为馏出物分离的任何剂。为此,分离hcfc-244bb作为馏出物,随后处理,以转化为hfo-1234yf。hcfo-1233xf任选从底部流纯化,并且可再循环,以转化为hcfc-244bb。根据本发明可用的萃取剂(特别是用于纯化馏出物中的hcfc-244bb)包括但不限于反式-hcfo-1233zd、顺式-hcfo-1233zd、hcfo-1223异构体和hcfo-1232异构体。在另一实施方案中,流可在后催化反应器中用过量的hf处理,直至其基本上不含任何未反应的hcfo-1233xf。在某些非限制性实施方案中,例如,hcfc-244bb流出物可在蒸气相催化反应器中处理,使得过量的hcfo-1233xf进一步转化为hcfc-244bb。这样的反应可在与通常使用相比温和的条件下进行。为此,可在低于通常用于蒸气反应的温度和压力条件下提供后催化反应。这样的条件已显示对于实现后反应流出物的几乎完全转化更理想。在一个实施方案中,后催化反应在约70℃-约120℃的温度下进行,或者在某些实施方案中,约100℃。其还可在约50psig-约120psig或约90psig的压力下进行。不必需要另外的hf使得反应进行,而是,可使用在初始反应中提供的过量的hf。然而,另外的hf可注入反应器至完成转化所需的程度。适用于hcfo-1233xf氟化为hcfc-244bb的任何催化剂为可接受的。这样的催化剂可包括在活性炭上担载的五氯化锑或氟化锑,或者本文确认的一种或多种催化剂,或者本领域另外已知的。在另一方面,使用共沸蒸馏可从hcfc-244bb进料流除去hcfo-1233xf。为此,可提供第三组分(例如但不限于hf),以形成共沸物或共沸组合物,将其随后从组合物分离。更具体地,加入第三组分与hcfo-1233xf和/或hcfc-244bb形成三元共沸物和/或二元共沸物。这些共沸物可使用标准分离手段(例如蒸馏)从溶液分离,使得一部分hcfc-244bb保留在溶液中。该部分随后分离为基本上不含hcfo-1233xf的纯化的hcfc-244bb。这样的共沸物和共沸分离或蒸馏的方法还可包括在美国专利号7,803,283和美国公布申请号2010/0187088和2009/0256110中公开的那些,各自的内容通过引用结合到本文中。在一个备选的实施方案中,将hcfc-244bb产物流光氯化,以使hcfo-1233xf饱和并形成hcfc-233ab(1,2,2-三氯-3,3,3-三氟丙烷),随后可从溶液除去。在某些实施方案中,光氯化反应包括在氯化试剂存在下,使hcfc-244bb进料流暴露于辐射。虽然不局限于此,在某些方面,辐射在紫外范围内,优选在约200-约400nm范围内。氯化剂可包括氯气,净的或具有稀释剂(例如氮气)。在uv光存在下,氯与hcfo-1233xf反应,使得在双键对向加入氯。反应优选在约-20℃至约200℃,甚至更优选约0℃-约100℃温度下在蒸气相中进行约5秒-约5小时,更优选约15秒-约30分钟的时间。反应产物(例如,hcfc-233ab)具有改变的沸点,因此其可随后经历一个或多个分离步骤(例如蒸馏),以将其从hcfc-244bb进料除去。举例说明蒸气相光氯化反应的前述实施方案不必局限于本发明。为此,还可使前述反应适于使用本领域已知的标准程序在液相中实施。在液相反应中,期望保持反应温度低于1233xf/244bb混合物的沸点。为此,可调节反应的压力或温度,使得反应混合物至少部分保留在液相中。前述蒸气相和液相反应和反应条件的实例在美国专利号7,205,444和5,336,377中提供,其内容通过引用结合到本文中。在甚至其它实施方案中,通过相对于hcfo-1233xf优先冷冻出或“冷捕集”hcfc-244bb,随后回收更纯的hcfc-244bb流,可分离hcfo-1233xf。更具体地,组合物的温度可降低至低于hcfc-244bb和hcfo-1233xf中的一个的凝固点,但是高于另一个的凝固点。在某些实施方案中,温度低于hcfc-244bb的凝固点,但是高于hcfo-1233xf的凝固点。这样的温度可为约-85℃至约-75℃,在其它实施方案中,约-81℃至约-78℃。结果是,以固体形式提供hcfc-244bb,而以液体形式提供hcfo-1233xf。固体hcfc-244bb可通过标准手段(例如,倾析、过滤、离心等)回收。在其它实施方案中,使用选自活性炭和沸石和/或分子筛的固体吸附剂,可除去hcfo-1233xf。更具体地,hcfo-1233xf和hcfc-244bb的混合物(作为液体或蒸气相)可通过选自活性炭和沸石和/或分子筛的固体吸附剂的固定床,其中所述沸石和/或分子筛的孔尺寸适于从hcfo-244bb选择性过滤hcfo-1233xf。在一种实施方案中,孔直径可在约3å-约10å范围内,在其它实施方案中,约3å-约5å。这样的分子筛的实施方案包括但不限于5a型和13x型分子筛,例如由uop、llc生产的那些。还可用的是5a合成沸石和天然衍生的沸石、菱沸石钙。前述不必然局限于本发明。为此,适合或可适合从hcfc-244bb选择性过滤hcfo-1233xf的任何分子筛和/或沸石可根据本发明使用。其中这样的分子筛和/或沸石可用于本发明的方法的实例在美国专利号4,906,796中提供,其内容通过引用结合到本文中。在甚至其它实施方案,hcfo-1233xf可通过标准层析法与hcfc-244bb分离。为此,在实现通过所述基材选择性保留一种组分和回收基本纯的另一组分的条件下,使hcfo-1233xf和hcfc-244bb的进料混合物与含有固定层析基材的床接触。作为一个实例,在60/80carbopackb载体上的5%fluocol相可用作这样的固定基材。这样的技术的实例可见于美国专利号3,903,187,其内容通过引用结合到本文中。在本发明的另一实施方案中,向包含2-氯-1,1,1,2-四氟丙烷和2-氯-3,3,3-三氟丙烯的组合物中加入液体氢氟酸。组合物可为得自上文描述的第二氟化过程的产物。在一种实施方案中,其浓度大于约0.01%重量并且低于约20%重量,在另一实施方案中,浓度为约0.05%-约10%重量,在另一实施方案中,氢氟酸的浓度低于约5%重量。所用的hcfc-244bb可来自前面步骤的反应器流出物或为其一部分。或者,其可从另一个来源引入。加入的量足以形成可分离出2-氯-1,1,1,2-四氟丙烷和形成产物的混合物,所述产物基本上不含2-氯-3,3,3-三氟丙烯,并且比起所得到的混合物,其2-氯-1,1,1,2-四氟丙烷更纯。在一种实施方案中,所得到的混合物不是三元共沸物或类似三元共沸的组合物。在另一实施方案中,hf存在于混合物中的hf量低于用于形成三元共沸物或类似共沸的组合物所需的量。在另一实施方案中,所得到的混合物形成三元共沸物或类似三元共沸的组合物。存在于混合物中的hf量低于混合物的10重量%。在另一实施方案中,其以低于混合物的约5重量%存在。在另一实施方案中,存在的量低于混合物的3重量%。将所得到的组合物蒸馏,收集基本上不含2-氯-3,3,3-三氟丙烯的产物。在一个实施方案中,利用与板式塔或盘式塔连通的再沸器。在该实施方案中,采用包含冷凝器和盘式塔的组件,连同具有通常用于再沸器的温度控制装置的再沸器。塔为塔板或塔盘的垂直组件,蒸气与液体在每一个塔板或塔盘上接触。将2-氯-1,1,1,2-四氟丙烷和2-氯-3,3,3-三氟丙烯的氢氟化溶液供料至引导到板式塔的进料。通常,进料位于板式塔的大约中部。在重力下液体向下流动经过塔,而蒸气在轻微压降的力下从塔板到塔板向上流动。然而,通过在再沸器中沸腾,产生最高压力,再沸器为位于底部与板式塔连通的加热器。蒸气经过每一个塔板中的开口并且接触跨塔板流动的液体。柱的再沸器部分用于除去在再沸器流中的组合物,该再沸器流包含基本上不含2-氯-3,3,3-三氟丙烯的2-氯-1,1,1,2-四氟丙烷,并且操作柱以在塔顶物流中除去包含2-氯-3,3,3-三氟丙烯(浓度比在再沸器部分中更大)和一些2-氯-1,1,1,2-四氟丙烷的混合物的组合物。采用与板式塔的上部连接的典型的回流冷凝器将后一种组合物(即,塔顶物部分)的一部分返回至板式塔,用于进一步蒸馏,并分离。当保持蒸馏时,从再沸器流连续除去包含基本上不含2-氯-3,3,3-三氟丙烯的更纯的2-氯-1,1,1,2-四氟丙烷的馏分。如上所述,前述分离技术可单独或以任何组合使用,以提供基本上不含hcfo-1233xf的hcfc-244bb的组合物。为此,分离技术可单独或以任何组合实施,导致具有低于约20.0重量%,低于约15.0重量%,低于约10.0重量%,低于约8.0重量%,低于约5.0重量%,低于约2.5重量%,或低于约1.0重量%hcfo-1233xf的组合物。在其它非限制性实施方案中,可实施这些技术,以提供完全不含hcfo-1233xf的组合物,或其中使用标准检测手段不能检测到hcfo-1233xf的量的组合物。例如,在一个非限制性实施方案中,混合物可使用共沸蒸馏然后光氯化然后分子筛/沸石的组合来处理,以降低hcfo-1233xf的水平,并且导致不含或基本上不含hcfo-1233xf的组合物。然而,这样的组合不局限于本发明,并且可适于结合本文提供的或另外的一种或多种任何技术,以实现期望水平的hcfo-1233xf。如上文描述的,本发明还提供基本上不含3,3,3-三氟丙炔的2-氯-1,1,1,2-四氟丙烷。因此,本发明的一个实施方案涉及制备基本上不含3,3,3-三氟丙炔的2,3,3,3-四氟丙烯的方法,所述方法包括:(i)提供包含基本上不含2-氯-3,3,3-三氟丙烯的2-氯-1,1,1,2-四氟丙烷的起始组合物;和(ii)使所述起始组合物与脱氢氯化催化剂接触,以生产包含2,3,3,3-四氟丙烯的最终组合物,所述最终组合物基本上不含3,3,3-三氟丙炔。本发明的另一方面涉及一种用于生产基本上不含3,3,3-三氟丙炔的2,3,3,3-四氟丙烯的方法,所述方法包括:(i)提供包括式i、ii或iii的化合物的起始组合物:cx2=ccl-ch2x (i);cx3-ccl=ch2 (ii);或cx3-chcl-ch2x (iii)其中x独立地选自f、cl、br和i,条件是至少一个x不是氟;(ii)使所述起始组合物与第一氟化剂接触,以生产包括2-氯-3,3,3-三氟丙烯和第一含氯副产物的第一中间组合物;(iii)使所述第一中间组合物与第二氟化剂接触,以生产包括2-氯-1,1,1,2-四氟丙烷和2-氯-3,3,3-三氟丙烯的第二中间组合物;(iv)降低在所述第二中间组合物中2-氯-3,3,3-三氟丙烯的水平,以生产包括2-氯-1,1,1,2-四氟丙烷并且基本上不含2-氯-3,3,3-三氟丙烯的第三中间组合物;和(v)使至少一部分2-氯-1,1,1,2-四氟丙烷脱氢氯化,以生产包括2,3,3,3-四氟丙烯的反应产物,所述反应产物基本上不含3,3,3-三氟丙炔。在一种实施方案中,形成的2,3,3,3-四氟丙烯基本上不含3,3,3-三氟丙炔。例如,形成为得自这些反应的产物的2,3,3,3-四氟丙烯含有低于约0.1重量%的3,3,3-三氟丙炔。在另一实施方案中,与2,3,3,3-四氟丙烯一起存在的3,3,3-三氟丙炔的浓度含有低于0.01重量%的3,3,3-三氟丙炔,在另一实施方案中,低于约0.005%重量。可通过本领域已知的技术(例如通过气相色谱法)测量与2,3,3,3-四氟丙烯一起存在的3,3,3-三氟丙炔的量。以下为本发明的实施例,并且不应解释为限制性的。实施例实施例1该实施例说明在hcfc-244bb连续蒸气相脱氢氯化为hfo-1234yf期间,在hcfc-244bb进料中hcfo-1233xf浓度如何影响hcfc-244bb的转化率。使用埋入3区域电炉中的3/4”直径的圆柱形inconel625反应器。使用放置在反应器内部的多点热电偶记录过程温度。将hcfc-244bb物料进料至垂直安装的反应器底部,在达到反应区域之前气化。在480℃、25psig和12g有机物/h的条件下进行反应。将流出物气体通过气体取样管,使得经由gc分析气体取样管的内含物,周期性监测反应的进程。如表1所示,随着在hcfc-244bb进料中hcfo-1233xf浓度从8.1降低至1.6gc面积%,hcfc-244bb的转化率显著提高。表1进料中的1233xfgc面积%244bb转化率,%20.021.014.021.68.121.64.628.31.636.4实施例2该实施例说明在hcfc-244bb连续蒸气相脱氢氯化反应为hfo-1234yf期间,在hcfc-244bb进料中hcfo-1233xf浓度如何影响hcfc-244bb的转化率。使用埋入3区域电炉中的3/4”直径的圆柱形inconel625反应器。使用放置在反应器内部的多点热电偶记录过程温度。将hcfc-244bb物料进料至垂直安装的反应器底部,在达到反应区域之前气化。在480℃、~25psig和12g有机物/h的条件下进行反应。将流出物气体通过气体取样管,使得经由gc分析气体取样管的内含物,周期性监测反应的进程。如图1所示,随着在hcfc-244bb进料中hcfo-1233xf浓度从1.6提高至20gc面积%,hcfc-244bb的转化率从超过40%显著降低至低于25%。实施例3该实施例说明在hcfc-244bb进料中hcfo-1233xf浓度如何影响在hcfc-244bb连续蒸气相脱氢氯化反应为hfo-1234yf期间形成的cf3cch的量。使用埋入sandbath炉中的1’’直径的u-形inconel625反应器。使用放置在反应器内部的多点热电偶记录过程温度。将hcfc-244bb物料进料至垂直安装的气化器的底部,在达到反应区域之前气化。在480℃、50psig和1lb有机物/h的条件下进行反应。将流出物气体周期性取样用于gc分析,以便监测反应的进程。如表2所示,随着在244bb进料中1233xf浓度从10.0降低至3.0gc面积%,形成的cf3cch的量和在反应器流出物中cf3cch/1234yf摩尔比显著降低。实施例4该实施例说明向244bb和1233xf的混合物中加入hf如何帮助在随后的蒸馏中除去1233xf。所用的蒸馏塔由10加仑再沸器、2英寸内径×8英尺propack柱和5平方英尺壳管式冷凝器组成。柱填充有monel1/4”pro-pack散堆填料,并且具有约30-35个理论塔板。蒸馏塔配备温度、压力和压差传感器。将106lb的3%hf/92.15%244bb/4.85%1233xf装入再沸器中。在40psig下开始间歇蒸馏。从柱塔顶物和再沸器周期性取出样品,通过gc分析有机组成,通过酸碱滴定分析hf浓度。当在再沸器中244bb浓度达到约98gc面积%(或1233xf为约2gc面积%)之后,开始连续蒸馏。包含hf、244bb和1233xf的混合进料作为液体形式通过位于塔中部的进料孔连续进料至塔内。将塔顶物流引向10重量%koh水溶液,用于酸去除,随后将有机物压缩,在通过干燥塔之后,收集到pcc(产物收集柱)中。将再沸器流直接压缩,在另一个pcc中收集。在操作过程期间,通过保持塔顶物取出速率和再沸器去除速率的总和等于进料速率,在再沸器中的液体液位保持在恒定的水平。如表3所示,1233xf在塔顶物流中富集,但是在再沸器流中贫乏。结果是,由蒸馏得到98gc面积%244bb。分析还指示包括在进料中的几乎所有的hf从塔顶物离开蒸馏塔(换言之,在再沸器流中不出现或出现很少hf)。实施例5如在实施例4中进行该蒸馏,使用相同的蒸馏塔,不同之处在于使用含有仅约400ppm的液体hf的进料。如表4所示,当存在仅数百ppm的hf时,在塔顶物取出流中1233xf浓度超过6gc面积%,而在再沸器排出流中它们保持在约2.5gc面积%,这指示存在即使仅数百ppm的hf也促进从244bb分离1233xf。根据本文描述的程序,将这样得到的244bb接下来脱氢氯化,以生产hfo-1234yf。实施例6在480℃下,随着在244bb中的1233xf浓度提高,3,3,3-三氟丙炔的形成增加在480℃下,以1.2ml液体/小时进料速率,将含有不同量的1233xf的244bb通过1/2”x12”(内径0.334”)inconel625管,用于脱氢氯化。通过gc和gc-ms分析来自反应器的流。测试结果在下表中列出。结果显示随着1233xf浓度提高,3,3,3-三氟丙炔的形成增加。表5实施例7在460℃下,随着在244bb中的1233xf浓度提高,3,3,3-三氟丙炔的形成增加在460℃下,以1.2ml液体/小时进料速率,将含有不同水平的1233xf的244bb通过1/2”x12”(内径0.334”)inconel625管,用于脱氢氯化。通过gc和gc-ms分析来自反应器的流。测试结果在下表中列出。结果显示随着1233xf浓度提高,3,3,3-三氟丙炔的形成增加。表6给出以上优选的实施方案和实施例以说明本发明的范围和精神。这些实施方案和实施例使得其它实施方案和实施例对于本领域技术人员显而易见。其它实施方案和实施例在本发明的预期内。因此,本发明应仅由所附权利要求限定。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1