单标记通用探针与特异性引物的组合物及其DNA检测方法与流程
本发明涉及实时荧光pcr检测技术领域,尤其涉及一种单标记通用探针与特异性引物的组合物及其dna检测方法。
背景技术:
在诸多实时荧光pcr检测技术中,以taqman探针技术应用最为广泛,该探针主要基于dna聚合酶的水解活性来引发探针上的荧光基团和淬灭基团不可逆的分离,进而形成荧光信号的累积。该技术虽然具有较高的检测效率,但是该技术仅能用于dna/rna分子实时分析定量,缺乏pcr后熔点分析的能力。因此,taqman探针技术应用过程中始终存在检测容量低的问题。
分子信标探针技术实质上是改良的taqman探针技术,即一种空间上呈“发夹”结构的taqman探针技术。探针中“发夹”结构的存在使得该技术能够应用于pcr后熔点分析。进一步来说,该技术一定程度上解决了pcr的检测容量的问题,但在检测效率和灵敏度不足使得其在实时分析中的应用受限。
相似的,双杂交探针由两条相邻的标记不同荧光基团的探针所组成,主要利用荧光基团间的荧光能量共振转移现象(fret)来实现荧光信号的检出,该技术同时具备了分子信标探针pcr后熔点分析以及taqman探针实时定量分析方面的优点。
公开号为cn102321765a的中国发明专利申请,公开了一种新的荧光pcr检测技术,该技术利用单标记探针同时具有引物和检测探针的特性,巧妙借助空间上的“茎环”结构实现荧光信号的可逆淬灭,同时具备了上述三种技术的优点,并且增加了形成“倒置”扩增曲线的反应特性,丰富了分析dna/rna分子的检测维度。
然而,以上四种实时荧光pcr检测技术均存在一个共同缺点,即需要针对特定的dna/rna分子设计特定探针,进一步来说,缺乏检测通用性,使得检测过程更加复杂,增加检测成本。
技术实现要素:
本发明的目的在于提供一种兼备上述四种技术的优点的新型检测工具和检测技术,即具备pcr实时分析检测和熔点分析能力,并能够在检测容量上进一步提升,同时克服现有的实时荧光pcr检测技术缺乏“检测通用性”的缺点的技术。
为实现上述目的,本发明的第一方面提供了一种基于单标记通用探针与特异性引物的组合物的dna检测方法,所述方法采用一组单标记通用探针和一组特异性引物构建pcr反应体系;所述单标记通用探针包括第一探针和第二探针,所述特异性引物包括上游引物和下游引物;
所述上游引物包括接头序列、第一标签序列和第一特异性序列,下游引物包括接头序列、第二标签序列和第二特异性序列,上游引物与下游引物的接头序列相同;所述第一特异性序列、第二特异性序列分别与靶标基因反义链、正义链的一段特异性序列反向互补;
所述第一探针包含第一通用序列,其一端标记有第一修饰基团,第二探针包含第二通用序列,其一端标记有第二修饰基团,所述第一修饰基团和第二修饰基团能配合发出荧光信号;所述第一通用序列与第一标签序列互补或反向互补,第二通用序列与第二标签序列相同或反向。
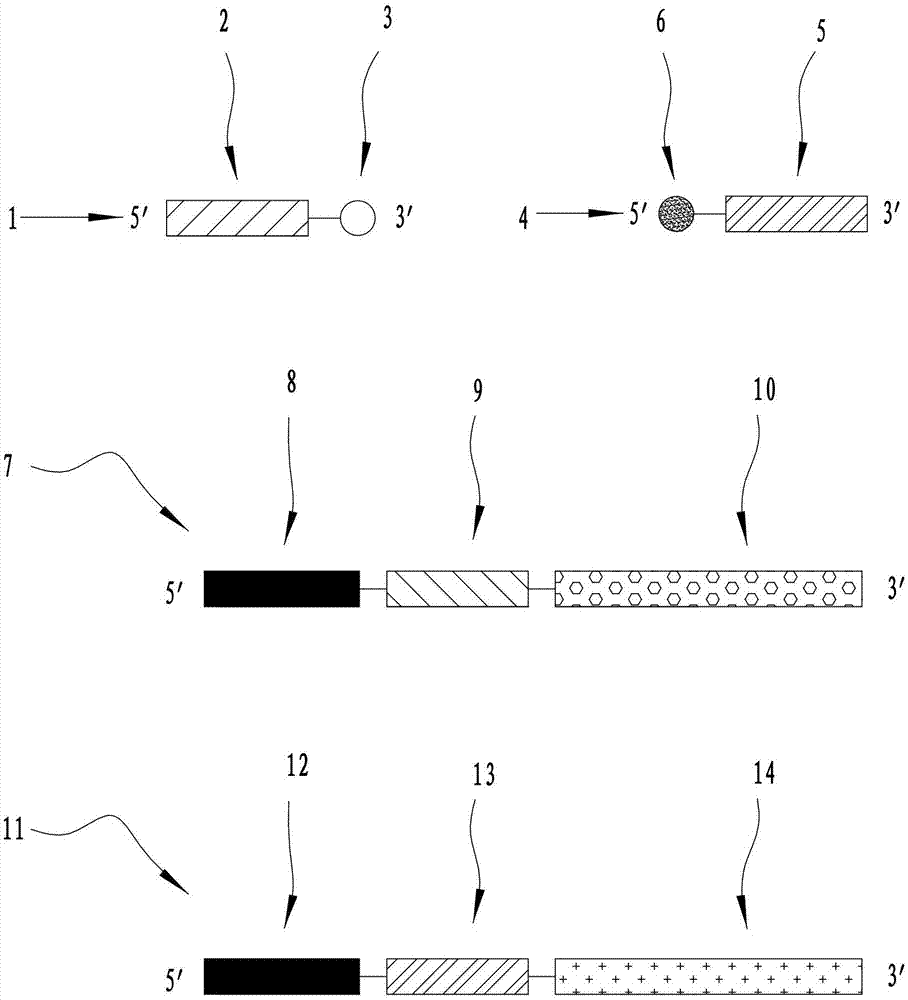
说明性地参照图1,图1中示出了单标记通用探针和特异性引物的结构示意图,其中第一探针1包括第一通用序列2和第一修饰基团3,第二探针4包括第二通用序列5和第二修饰基团6。上游引物7包括接头序列8、第一标签序列9和第一特异性序列10,下游引物11包括接头序列12、第二标签序列13和第二特异性序列14。
示例性的,在图1中,第一通用序列2与第一标签序列9反向互补,第一修饰基团3标记于第一探针1的3′端;第二通用序列5与第二标签序列13相同,第二修饰基团6标记于第二探针4的5′端。
运用以上的探针和引物构建反应体系,参照图2a-2d,其以待测靶标基因的正义链15为示例,示出了其进行pcr扩增反应前三轮的扩增原理示意图。相应的,待测靶标基因的反义链的扩增情况可以参照该正义链的扩增情况。在图2a中示出了一轮扩增反应的情况,待测靶标基因变性解链为正义链15后,其上的特异性序列16与下游引物11的第二特异性序列14反向互补,在后续的退火和延伸过程中,下游引物11与正义链15结合,并沿其自身5′-3′的方向延伸,形成一轮扩增的pcr产物的单链17,其相当于下游引物11与对应的反义链的组合。
接下来参照图2b,其示出了二轮扩增反应的情况,单链17由于具有反义链上的特异性序列(位于其3′端,未标示),其与上游引物7的第一特异性序列10反向互补,因而在退火和延伸过程中,上游引物7与单链17结合,并沿其自身5′-3′的方向延伸,形成二轮扩增的pcr产物的单链18,其相当于上游引物7、正义链15、下游引物11的反向互补序列的组合,此时,请参照图2c。
最后,在三轮扩增反应中,参照图2d,二轮扩增的pcr产物经变性后解为单链18,在单链18中,由于接头序列12经过前二轮扩增后,形成为原始序列的反向互补序列12′,因而在退火时,与接头序列12序列相同的接头序列8与该反向互补序列12′更倾向于相互杂交,使得单链18自身形成一茎环结构体19。
此外,单链18上具有与对应下游引物11的第二标签序列13的反向互补序列13′,且第二探针4的第二通用序列5与第二标签序列13相同,即第二探针4的第二通用序列5与该反向互补序列13′反向互补,而第一探针1的第一通用序列2本身就与单链18上所具有的第一标签序列9反向互补,这样在退火过程中,第一探针1和第二探针4由原来的游离状态演变为与茎环结构体19结合,使得第一修饰基团3和第二修饰基团6靠近。在这里,第一探针1和第二探针4应当是过量的,以保证有足够的单标记通用探针与该茎环结构体19结合。
图示实施例的第一修饰基团和第二修饰基团可为荧光基团和淬灭基团,如此一来,当单标记通用探针处于游离状态时,淬灭基团无法对荧光基团进行淬灭,整个pcr混合液中存在较高荧光信号本底。随着第一探针1和第二探针4与茎环结构体19结合,第一修饰基团和第二修饰基团靠近,荧光基团被淬灭基团所淬灭,随着pcr循环数的增加,茎环结构体19不断积累,pcr混合液荧光信号本底随循环次数增加而下降,最终呈现倒置的扩增曲线。
在下述提及到的可能替代的实施方式中,第一修饰基团和第二修饰基团为受体荧光基团或者供体荧光基团,则出现相反的荧光情况。随着第一探针1和第二探针4与茎环结构体19结合,第一修饰基团和第二修饰基团靠近,其中的供体荧光基团的发射谱与受体荧光基团的激发谱具有一定程度的重叠,当供体荧光基团被激发时,受体荧光基团会因供体荧光基团激发能的转移而被激发,表现为供体荧光基团产生的荧光强度较其单独存在时要低的多,而受体荧光基团发射的荧光信号却大大增强。因而,随着pcr循环数的增加,茎环结构体19不断积累,pcr混合液荧光信号本底随循环次数增加而上升,最终呈现正置的扩增曲线。
因而,从上述说明可以得到,本发明的图示实施例(1)探针只需要单标记,且设计的探针序列为通用序列,其与特异性引物的对应部分相同或互补,在设计探针序列时省时省力,节约成本;(2)只有当pcr产物形成正确的二级茎环结构,且探针特异性杂交时才能出现信号差,特异性高,选择性好;(3)可用于熔解分析,可对检测靶序列定性分析;(4)可灵活使用各种荧光原理不同的修饰基团,从而形成“倒置”及“正置”的扩增曲线。
在某一可能的实施方式中:所述接头序列、第一标签序列、第二标签序列为任意的不与靶标基因杂交的附加核酸序列,从而避免不必要的分子内自杂交和扩增。
在以上说明中可以看到,为使第一修饰基团3和第二修饰基团6在单标记通用探针与茎环结构体19结合时的距离较近,可替代地配合调整第一通用序列2、第二通用序列5的方向以及第一修饰基团3、第二修饰基团6的标记位置,使得单标记通用探针与茎环结构体19结合时,第一修饰基团3和第二修饰基团6始终位于较近的距离内,提高了荧光信号检测的精确度。
相应的,对于第一探针,在某一可能的实施方式中:当所述第一通用序列与第一标签序列互补,所述第一修饰基团标记于第一探针的5′端;当所述第一通用序列与第一标签序列反向互补,所述第一修饰基团标记于第一探针的3′端。
相应的,对于第二探针,在某一可能的实施方式中:当所述第二通用序列与第二标签序列相同,所述第二修饰基团标记于第二探针的5′端;当所述第二通用序列与第二标签序列反向,所述第二修饰基团标记于第二探针的3′端。
如上说明的,在某一可能的实施方式中:所述第一修饰基团和第二修饰基为受体荧光基团或者供体荧光基团,以形成正置的扩增曲线。
如上说明的,在某一可能的实施方式中:所述第一修饰基团和第二修饰基团为荧光基团和淬灭基团,以形成倒置的扩增曲线。
在某一可能的实施方式中:所述接头序列的tm值范围在50-65℃;所述单标记通用探针序列的tm值范围在50-65℃;
所述第一特异性序列、第二特异性序列、接头序列以及单标记通用探针序列的tm值相差不超过5℃。
在某一可能的实施方式中:所述方法包括以下步骤:合成所述单标记通用探针、特异性引物;构建pcr反应体系,对待测靶标基因进行pcr扩增;在pcr扩增反应结束后,收集pcr产物的荧光信号以形成相应的扩增曲线,对待测靶标基因进行分析。
如上说明的,在某一可能的实施方式中:所述pcr扩增反应至少进行三轮扩增;进行第三轮扩增时,二轮扩增的pcr产物经过变性、退火后,其单链自身形成一茎环结构体;
所述单链两端对应所述接头序列的部分杂交,构成该茎环结构体的锁合部;所述单链两端对应所述第一标签序列、第二标签序列的部分构成探针杂交部,该探针杂交部与锁合部相邻;
所述单标记通用探针的第一探针和第二探针分别与探针杂交部上对应的互补序列杂交,第一修饰基团和第二修饰基团靠近。
为实现上述目的,本发明的第二方面提供了一种单标记通用探针及特异性引物的组合物,其用于dna检测中构建pcr反应体系,所述组合物包括一组单标记通用探针和一组特异性引物,所述单标记通用探针和特异性引物为上述技术方案中所述的单标记通用探针和特异性引物。
本发明所提供的单标记通用探针与特异性引物的组合物及其dna检测方法,采用一组特异性引物和一组单标记通用探针,通过使pcr产物形成正确的二级茎环结构后,特异性单标记通用探针与该二级茎环结构杂交,使得标记于该组单标记通用探针的两修饰基团相互靠近,以与游离状态时的荧光信号产生一定量的信号差,从而具备高特异性。通过改变修饰基团的荧光原理种类,可以得到“倒置”和“正置”的扩增曲线和熔解曲线,且该单标记通用探针的序列具备通用性,能够达到用相同的探针检测不同的靶标基因的目的,该检测方法能够应用于荧光定量pcr、多重检测等等,大大地节约物质和人力成本。
附图说明
图1示出了本发明实施例的单标记通用探针和特异性引物的结构示意图;
图2a-2d示出了本发明实施例的dna检测方法的原理示意图,其中,图2a示出了一轮扩增反应的情况,图2b示出了二轮扩增反应的情况,图2c示出了二轮扩增反应的pcr产物的单链结构,图2d示出了三轮扩增反应的情况;
图3a示出了实施例一梯度稀释样本的扩增曲线;
图3b示出了实施例一梯度稀释样本扩增曲线的ct值与dna起始拷贝数的对数关系曲线;
图3c示出了实施例一梯度稀释样本的熔解曲线图;
图4a示出了实施例二abcb1基因样本及abcc1基因样本的熔解曲线图;
图4b示出了实施例二gstp1基因样本的熔解曲线图;
图4c示出了实施例二mvp基因样本的熔解曲线图。
具体实施方式
以下结合附图及具体实施例对本发明作进一步的说明。值得说明的是,以下具体实施例的举例是为了方便本领域技术人员进一步了解本发明的实施过程所进行的描述,不能理解为对本发明精神具体化的限制。实施例中未注明具体技术或条件的,参考j.萨姆布鲁克等著,黄培堂等译的《分子克隆实验指南》(第四版,科学出版社)或者按照产品说明书进行,所使用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例中使用仪器:实时荧光pcr仪(lightcylcer480ⅱ,瑞士罗氏公司),紫外可见分光光度计(nd-1000,美国nanodrop公司),微量台式离心机(相仪,h1650-w),以下实施例中所有序列的合成均来自生工生物工程(上海)有限公司,所使用的基因组dna样本来源于正常人的外周血提取,均采用康为世纪生物科技公司的bloodgenomicdnaminikit,并遵照其说明书的提取方式提取获得。外周血样品由厦门市中山医院提供,标本的使用均获得当事人或其监护人的许可。
实施例一
选择人基因组上的序列,基因登录号为al354732.30,依据以上所述的原则及说明,设计并合成相应的引物及探针,其序列如下:
上游引物:
5′-tgcagggtcgctaagaagt-gttttcccagtcacgacac-gtcagtgtaaggaataacgatgac-3′
下游引物:
5′-tgcagggtcgctaagaagt-ctgaacagctatgaccatgcaa-ttttgtcgaattgctgtgatg-3′
第一探针:5′-gtcgtgactgggaaaac-bhq-3′
第二探针:5′-rox-ctgaacagctatgaccatgcaa-3′
其中上游引物中,由5′端至3′端,依次用分隔线“-”隔开了各组成部分,其依次为接头序列、第一标签序列和第一特异性序列;对应的,在下游引物中,依次为接头序列、第二标签序列和第二特异性序列。
第一探针的3′端标记bhq淬灭基团,第二探针的5′端标记rox荧光基团,它们与扩增产物的茎环结构体杂交形成复合体,在形成复合体后发生荧光能量共振转移从而达到实时监测产物形成的目的,随着pcr循环数的增加,体系中的荧光信号逐渐降低形成倒置的扩增曲线,如图3a所示。
具体的,pcr反应体系中,选择用量为8.46×106copies/ul、8.46×105copies/ul、8.46×104copies/ul、8.46×103copies/ul、8.46×102copies/ul、8.46×101copies/ul(即10倍梯度稀释)的人工构建的人类基因质粒标准品作为反应模板。10μlpcr反应体系内含2μl人基因组模板,75mmol/ltris-hcl,其ph=9.0,20mmol/l(nh4)2so4,0.01%tween20,50mmol/lkcl,0.4utaq酶,2.5mmol/lmg2+,0.1μmol/l第一探针,0.1μmol/l第二探针,0.4μmol/l上、下游引物。pcr反应程序为:95℃5分钟;95℃10秒,60℃20秒,72℃20秒,45个循环;60℃退火阶段采集荧光信号。熔解程序为:95℃1min,55℃1min,53℃-80℃melt。
如图3a所示,10倍梯度稀释样本的扩增曲线1a体现较好的梯度,而2a(阴性对照,没有模板的存在,即为ddh2o)则无特异产物的扩增,故检测不到特异信号。8.46×106copies/ul模板用量的反应管的ct值为18.53(ct值的含义是:每个反应管内的荧光信号达到设定的域值时所经历的循环数)、8.46×105copies/ul模板用量的反应管的ct值为22.23、8.46×104copies/ul模板用量的反应管的ct值为25.93、8.46×103copies/ul模板用量的反应管的ct值为31.30、8.46×102copies/ul模板用量的反应管的ct值为34.82、8.46×101copies/ul模板用量的反应管的ct值为37.92。可见,相邻梯度模板用量的ct值差接近3.3(理论上,扩增效率为100%时,模板用量差异10倍时ct值差为3.3),结果表明该体系具有较高的反应效率。在图3b中,也体现出梯度样本扩增曲线的ct值与其dna起始拷贝数的对数有很好的线性关系(r2=0.9935),体现该技术较强的定量检测能力。如图3c所示,10倍梯度稀释样本的熔解曲线1a所在的位置的tm值为64.5℃,是为特异的熔解曲线,说明pcr的扩增产物为特异的拟检测基因的产物,而熔解曲线2a(阴性对照,没有模板的存在,即为ddh2o)则无特异产物的扩增,故检测不到特异的熔解曲线。
实施例二
本实施例以4种人类的耐药基因,分别为abcb1基因(geneid:5243)、abcc1基因(geneid:4363)、gstp1基因(geneid:2950)、mvp基因(geneid:5243)为研究对象,并建立多重pcr反应体系,针对以上4种耐药基因设计引物和探针如下:
对于abcb1基因,所设计的引物及探针的序列为:
abcb1-f:
5′-tgcagggtcgctaagaagt-gttttcccagtcacgac-gtctacagttcgtaatgctg-3′
abcb1-r:
5′-tgcagggtcgctaagaagt-ctgaacagctatgaccatgcaa-gacaagtttgaagtaaatgcc-3′abcb1-p1:5′-gtcgtgactgggaaaac-bhq-3′
abcb1-p2:5′-fam-ctgaacagctatgaccatgcaa-3′
对于abcc1基因,所设计的引物及探针的序列为:
abcc1-f:
5′-tgcagggtcgctaagaagt-gttttcccagtcacgac-cagatgacacctctcaacaa-3′
abcc1-r:
5′-tgcagggtcgctaagaagt-ctgaacagctatgaccatg-tccctgaagactgaactcc-3′
abcc1-p1:5′-gtcgtgactgggaaaac-bhq-3′
abcc1-p2:5′-fam-ctgaacagctatgaccatg-3′
对于gstp1基因,所设计的引物及探针的序列为:
gstp1-f:
5′-tgcagggtcgctaagaagt-ccactgtctgaggcttga-cggagacctcaccctgta-3′
gstp1-r:
5′-tgcagggtcgctaagaagt-agagctcgtttagtgaaccg-ccttcccatagagccca-3′
gstp1-p1:5′-tcaagcctcagacagtgg-bhq-3′
gstp1-p2:5′-hex-agagctcgtttagtgaaccg-3′
对于mvp基因,所设计的引物及探针的序列为:
mvp-f:
5′-tgcagggtcgctaagaagt-ccactgtctgaggcttga-caacactgccctccatc-3′
mvp-r:
5′-tgcagggtcgctaagaagt-agagctcgtttagtgaac-gcctgaatgatctccacga-3′
mvp-p1:5′-tcaagcctcagacagtgg-bhq-3′
mvp-p2:5′-rox-agagctcgtttagtgaac-3′
关于上述探针和引物的说明,可参照实施例一,此处不再赘述。
本实施例的pcr反应体系中,10μlpcr反应体系内含2μl人基因组模板,75mmol/ltris-hcl,其ph=9.0,20mmol/l(nh4)2so4,0.01%tween20,50mmol/lkcl,0.4utaq酶,2.5mmol/lmg2+,各探针的终浓度为0.1μmol/l,各引物的终浓度为0.3μmol/l。pcr反应程序为:95℃5分钟;95℃10秒,60℃20秒,72℃20秒,45个循环;60℃退火阶段采集荧光信号。熔解程序:95℃1min,50℃1min,50℃-80℃melt。
本实施例所构建的多重pcr反应体系可以同时检测4种耐药基因,其检测结果如图4a、4b、4c结果所示,为在建立的多重pcr反应体系中检测人类基因组的模板的检测结果。
图4a示出了abcb1基因样本及abcc1基因样本的熔解曲线图,由于相应的探针均标记了fam荧光基团,故在fam通道中,abcb1基因对应的熔解曲线1b的特异tm值为56.2℃,abcc1基因对应的熔解曲线1b的特异tm值为69.2℃,而曲线2b(阴性对照,没有模板的存在,即为ddh2o)无特异性产物的扩增,故检测不到特异信号。图4b示出了gstp1基因样本的熔解曲线图,在hex通道中,gstp1基因对应的熔解曲线1c的特异tm值为66.2℃,曲线2c(阴性对照,没有模板的存在,即为ddh2o)无特异性产物的扩增,故检测不到特异信号。图4c示出了mvp基因样本的熔解曲线图,在rox通道中,mvp基因对应的熔解曲线1d的特异tm值为58.6℃,曲线2d(阴性对照,没有模板的存在,即为ddh2o)无特异性产物的扩增,故检测不到特异信号。
以上所述仅为本发明的优选实施例,并非因此限制其专利范围,凡是利用本发明说明书及附图内容所作的等效结构变换,直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!
- 用于检测AEDs引起的SJS/TEN的引物、试剂盒及方法与制造工艺
- 检测植原体的LAMP引物组及其试剂盒和应用的制造方法与工艺
- 用于多重PCR检测致腹泻寄生虫的引物组及试剂盒的制造方法与工艺
- 使用重叠非等位基因特异性引物和等位基因特异性阻断剂寡核苷酸的组合物进行等位基因特异性扩增的制造方法与工艺
- 一种基于特异性SS-COI引物的丽草蛉PCR快速检测方法与制造工艺
- 一种利用线粒体D-loop区特异区域鉴定鼋的标记引物及方法
- 一种用于检测大豆拟茎点种腐病菌的特异性lamp引物组合物及其应用
- 一种dna凝胶切胶装置的制造方法
- 一种用于dgge分析混合中药粉末组成物种的引物的制作方法
- 一种用于dgge鉴定混合中药粉末组成物种的引物的制作方法
- 玉米低镁条件下特异性转运镁离子基因ZmMGT10及其应用的制造方法与工艺
- CRISPR‑Cas9特异性敲除猪SLA‑3基因的方法及用于特异性靶向SLA‑3基因的sgRNA与制造工艺
- 毛酸浆的分子特异性标记引物及鉴别方法与制造工艺
- 一种检测CYP2C9基因多态性的ARMS引物的制造方法与工艺
- 构建在海马体区域特异性敲除IKKα基因的小鼠模型的方法及打靶载体和试剂盒与制造工艺
- 使用重叠非等位基因特异性引物和等位基因特异性阻断剂寡核苷酸的组合物进行等位基因特异性扩增的制造方法与工艺
- 一种利用线粒体D-loop区特异区域鉴定鼋的标记引物及方法
- 一种用于检测大豆拟茎点种腐病菌的特异性lamp引物组合物及其应用
- 一种针对膨胀污泥中Eikelboom Type 0092型丝状菌群特异性引物和方法
- 脯氨酸特异性蛋白酶基因及其应用