具有短侧链的苯并二氢吡喃-6-醇的制备方法与流程
技术实现要素:
本发明涉及一种制备式(iii)化合物的方法,
包括使式(i)的化合物与式(iia)、(iib)或(iic)的化合物在酸催化剂的存在下并且在两种溶剂的混合物中反应的步骤,
其中or是oh、乙酸酯、甲酸酯、丙酸酯、丁酸酯或苯甲酸酯,
a是ch2,
r1是c1-5-烷基,
r2是h或c1-2-烷基,
r4是h或c1-4-烷氧基或c1-4-烷基,
r3和r5彼此独立地为h或c1-4-烷基,并且
两种溶剂中的第一种是水,
两种溶剂中的第二种选自脂族c5-8烃、脂环族c5-8烃、二烷基醚和甲基取代的苯及其任何混合物。
背景技术:
q.wang、x.she、x.ren、j.ma、x.pan描述了于80%甲酸中2-甲基丁-3-烯-2-醇与1,4-氢醌(“hq”)缩合为2,2-二甲基苯并吡喃-6-醇(“dmc”;式(iii-1)化合物),产率为56%(“thefirstasymmetrictotalsynthesisofseveral3,4-dihydroxy-2,2-dimethyl-chromanderivatives(若干3,4-二羟基-2,2-二甲基-苯并二氢吡喃衍生物的首次不对称全合成)”,tetrahedron:asymmetry,2004年,第15卷第1期,第29-34页)。
还已经描述了使用三氟乙酸作为反应介质(dmc的产率为33%)的反应(参见f.m.d.ismail,m.j.hilton,
或者,也可以根据各种出版物使用异戊二烯进行反应,产率在53-62%之间(参见r.h.cichewicz,v.a.kenyon,s.whitman,n.m.morales,j.f.arguello,t.r.holman,p.crews:“redoxinactivationofhuman15-lipoxygenasebymarine-derivedmeroditerpenesandsyntheticchromanes:archetypesforauniqueclassofselectiveandrecyclableinhibitors”(海洋衍生的二萜和合成苯并二氢吡喃对人类15-脂氧合酶的氧化还原灭活作用:独特类型的选择性和可循环利用抑制剂的原型),j.am.chem.soc.2004,126,14910–14920;g.p.kalena,a.jain,a.banerji:<<amberlyst15catalyzedprenylationofphenols:one-stepsynthesisofbenzopyrans.”(amberlyst15催化苯酚的烯丙基化:苯并吡喃的一步合成),molecules1997,2,100–105(使用amberlyst15在thf/庚烷中,65-70℃,2.5h,61%);v.k.ahluwalia,k.k.arora,r.s.jolly:“acid-catalysedcondensationofisoprenewithphenols.formationof2,2-dimethylchromans.”(异戊二烯与酚的酸催化缩合.2,2-二甲基苯并吡喃的形成),j.chem.soc.,perkintrans.11982,335–338(h3po4);f.bigi,s.carloni,r.maggi,c.muchetti,m.rastelli,g.sartori:“reactionbetweenphenolsandisopreneunderzeolitecatalysis.highlyselectivesynthesisofchromansando-isopentenylphenols.”(沸石催化下的苯酚与异戊二烯之间的反应。苯并二氢吡喃和邻异戊烯基酚的高度选择性合成),synthesis1998,301–304(沸石hsz-360,高压釜,120℃,5h,产率50%);r.nast,m.dahm,k.ley(bayer):“polyurethanefoamsstabilizedwith6-hydroxychromans”(用6-羟基苯并二氢吡喃稳定的聚氨酯泡沫),de1945212(h3po4,在二甲苯/石油醚中,产率62%)。
此外,已显示出乙酸异戊烯酯(prenylacetate)与in(otf)3(稀土金属铟的盐)催化的hq反应(参见v.vece,j.ricci,s.poulain-martini,p.nava,y.carissan,s.humbel,
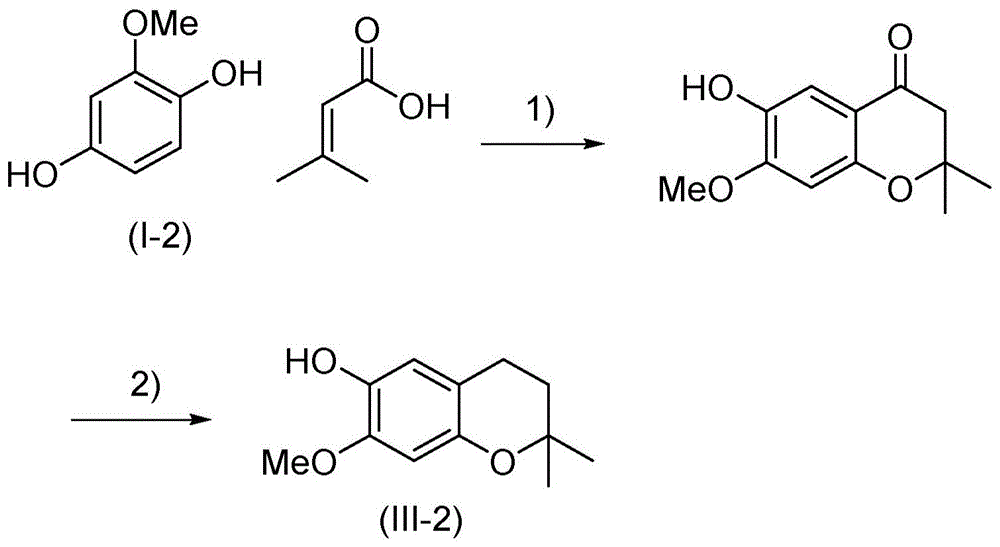
文献中已经描述了式(iii-2)的化合物的两步合成,反应方案如图1所示。
步骤1):以meso3h为溶剂;50mol%的p2o5,产率91%(参见f.camps,j.coll,a.messeguer,mapericás,s.ricart,wsbowers,dmsoderlund:“animprovedprocedureforthepreparationof2,2-dimethyl-4-chromanones.”(一种改进的制备2,2-二甲基-4-苯并二氢吡喃酮的方法),synthesis,1980年第725-727页。
步骤2):lialh4,et2o,产率为87%(参见p.anastasis,p.e.brown:“analoguesofantijuvenilehormones.”(抗幼激素的类似物),j.chem.soc.,perkintrans.11982,2013–2018.)。
这两个步骤的总产率为79%。
然而,当前由文献已知的方法提供的产率对于可持续的工业方法而言还不够高。因此,需要提供一种基于式(i)的化合物以高产率制造式(iii)的化合物的可持续的工业方法,其中避免使用卤化溶剂。
发明详述
因此,本发明满足了这种需要,本发明涉及制备式(iii)化合物的方法,
包括使式(i)的化合物与式(iia)、(iib)或(iic)的化合物在酸催化剂的存在下并且在两种溶剂的混合物中反应的步骤,
其中or是oh、乙酸酯、甲酸酯、丙酸酯、丁酸酯或苯甲酸酯,
a是ch2,
r1是c1-5-烷基,
r2是h或c1-2-烷基,
r4是h或c1-4-烷氧基或c1-4-烷基,
r3和r5彼此独立地为h或c1-4-烷基,并且
两种溶剂中的第一种是水,
两种溶剂中的第二种选自脂族c5-8烃、脂环族c5-8烃、二烷基醚和甲基取代的苯及其任何混合物。
因此,避免了卤化溶剂的使用。有利地,仅通过将产物相与含催化剂和过量的式(i)化合物的相分离,就可以重复使用所用的酸催化剂和过量的式(i)化合物。
原料和产品
or优选地为oh或乙酸酯。
r1优选地为甲基。
r2优选为h或甲基;更优选地r2为甲基。
r4优选为h或甲氧基或甲基,更优选地r4为h或甲氧基。
r3和r5优选地彼此独立地为h或甲基。
在本发明的上下文中,“烷基”和“烷氧基”分别包括直链烷基和支链烷基,以及直链烷氧基和支链烷氧基。
图2-5示出了用作原料的最优选化合物和通过本发明方法获得的作为产物的最优选化合物。
图2示出了由1,4-氢醌(式(i-1)的化合物)和2-甲基丁-3-烯-2-醇(式(iia-1)的化合物)开始的2,2-二甲基苯并吡喃-2-醇(式(iii-1)的化合物)的合成。
图3示出了由1,4-氢醌(式(i-1)的化合物)和异戊二烯(式(iib-1)的化合物)开始的2,2-二甲基苯并吡喃-2-醇(式(iii-1)的化合物)的合成。
图4示出了由1,4-氢醌(式(i-1)的化合物)和异戊烯基(prenyl)衍生物(式(iic-1)的化合物)开始的2,2-二甲基苯并吡喃-2-醇(式(iii-1)的化合物)合成)的合成。
图5示出了由1,4-氢醌(式(i-1)的化合物)和异戊烯醇(prenol)(式(iic-2)的化合物)开始的2,2-二甲基苯并吡喃-2-醇(式(iii-1)的化合物)的合成。
在本发明的一个优选实施方式中,式(i)的化合物与式(iia)、(iib)或(iic)的化合物的摩尔比在6.0:1-1.1:1的范围内,更优选在4.0:1至1.3:1的范围内,甚至更优选在3.0:1至1.5:1的范围内,最优选在2.5:1至1.7:1的范围内。
在本发明的另一个实施方式中,实现了关于上述原料和优选选项的本发明的所有实施方式。
溶剂混合物
脂族c5-8烃的优选实例是己烷和庚烷。术语“己烷”涵盖了正己烷以及己烷的异构体的任何混合物。同样适用于庚烷。
脂环族c5-8烃的优选实例是环己烷。
二烷基醚中的烷基可以是相同的或不同的,优选地它们是不同的。优选地,烷基是脂族直链c1-5-烷基或支链c3-6烷基。二烷基醚的优选实例是甲基叔丁基醚。
甲基取代的苯的优选实例是邻二甲苯、间二甲苯、对二甲苯、均三甲苯、假枯烯和甲苯。
优选地,第二溶剂是己烷、环己烷、庚烷、邻二甲苯、间二甲苯、对二甲苯、均三甲苯、假枯烯、甲基叔丁基醚或甲苯,及其任何混合物。更优选地,第二溶剂是己烷、环己烷、庚烷、邻二甲苯、间二甲苯、对二甲苯、均三甲苯、假枯烯、甲基叔丁基醚或甲苯。
在本发明的一个优选实施方式中,两种溶剂中的第一种是水,而两种溶剂中的第二种选自均三甲苯、假枯烯、邻二甲苯、间二甲苯、对二甲苯或甲苯,更优选地两种溶剂中的第一种是水,而两种溶剂中的第二种选自邻二甲苯、间二甲苯、对二甲苯或甲苯,最优选地,两种溶剂中的第一种是水,而两种溶剂中的第二种是甲苯。
在本发明的一个优选实施方式中,反应期间第一溶剂与第二溶剂的体积比在1:4至4:1的范围内,更优选地第一溶剂与第二溶剂的体积比在1:3至3:1的范围内,最优选第一溶剂与第二溶剂的体积比在1:2至2:1的范围内。
在本发明的另一个实施方式中,两种溶剂的总量在1至8kg每千克式(i)化合物的范围内,优选在2至6kg每千克式(i)化合物的范围内,更优选在2.5至5.5kg每千克式(i)化合物的范围内。
在本发明的另一个实施方式中,实现了关于溶剂和如上所述的优选选项的本发明的所有实施方式。
水可以原样添加至反应混合物中或作为酸催化剂的水溶液添加,其优选如下所述,即,可以使用水和酸催化剂的混合物。在本发明的一个优选的实施方式中,以上给出的水的优选量还包括酸催化剂的水溶液中所含的水。
酸催化剂
合适的酸催化剂的实例是布朗斯台德酸和路易斯酸及其任何混合物。
布朗斯台德酸的实例是硫酸、磷酸、酸性离子交换树脂(例如amberlyst15)、酸性粘土(例如蒙脱土k-10)、沸石(例如hsz-360)、盐酸、三氟乙酸、三氯乙酸、乙酸、甲酸、甲磺酸、苯磺酸、对甲苯磺酸、乙磺酸、三氟甲磺酸、双(全氟烷基-磺酰基)甲烷(r'so2)(r”so2)ch2(其中r'和r”各自独立地表示式cnf2n+1的全氟烷基,其中n是1至10的整数)、三(全氟磺酰基)甲烷(r'so2)(r“so2)(r″′so2)ch(其中r'、r″和r″′彼此独立地表示式cnf2n+1的全氟烷基,其中n是1至10的整数,并且其中r′、r″和r″′中的至少两个是相同的全氟烷基,或r'表示五氟-苯基(-c6f5),r”和r″′各自表示相同的上式cnf2n+1全氟烷基)、甲烷三磺酸和双(三氟甲基-磺酰基)酰亚胺,以及它们的任何混合物,其中优选使用单一催化剂。
路易斯酸的实例为sc(otf)3、sc(ntf2)3、sccl3、yb(otf)3、ybcl3、cu(otf)2、fecl2、fe(otf)2、zncl2、zn(otf)2、zn(ntf2)3、ycl3、y(otf)3、incl3、inbr3、in(otf)3、in(ntf2)3、la(otf)3、ce(otf)3、sm(otf)3、gd(otf)3、bi(otf)3在不存在或存在2,2-联吡啶的情况下及其任何混合物,其中优选使用单一催化剂。
优选地,酸催化剂是硫酸、盐酸、甲酸、amberlyst15、三氟乙酸或磷酸或其水溶液/悬浮液,更优选地,酸催化剂是硫酸、盐酸或amberlyst15,最优选地,酸催化剂是硫酸。也可以使用这些催化剂的混合物。
在本发明的一个优选实施方式中,酸催化剂是硫酸,并且所述硫酸的浓度在0.1至10mol/l的范围内,优选其中所述硫酸的浓度在0.4至4.0mol/l的范围内。
在本发明的另一个优选实施方式中,相对于式(iia)、(iib)或(iic)的化合物的量,酸催化剂的量在0.01至10摩尔当量的范围内,更优选在0.05至5摩尔当量的范围内,最优选在0.1至1摩尔当量的范围内。
在本发明的另一个实施方式中,实现了关于上述催化剂和优选选项的本发明的所有实施方式。
反应条件
反应优选在50至140℃范围内的温度下进行,更优选在60至120℃范围内的温度下,甚至更优选在70至100℃范围内的温度下,最优选在75至90℃范围内的温度下进行。
反应优选在0.5至20巴(绝对)范围内的压力下进行,更优选在0.7至10巴(绝对)范围内的压力下,最优选在0.8至5巴(绝对)范围内的压力下进行。
在根据本发明的一个方法中,酸催化剂是可再循环的(=可重复使用的),这是本发明的另一个优点。
在本发明的另一个实施方式中,实现了关于上述反应条件和优选选项的本发明的所有实施方式。
在本发明的最优选的实施方式中,实现了关于原料、溶剂、催化剂和反应条件(包括如上所述的优选选项)的本发明的所有实施方式。
现在在以下非限制性实施例中进一步说明本发明。
实施例
比较例:
实施例1:从1,4-氢醌和2-甲基丁-3-烯-2-醇开始合成2,2-二甲基苯并吡喃-2-醇
在室温下,在装有回流冷凝器、磁力搅拌器和氩气供应的500毫升烧瓶中装入18.6克氢醌(167毫摩尔,99%,2.0摩尔当量),并将其溶于250克甲酸(5.4摩尔,65摩尔当量)和70克水中。
加入7.2g的2-甲基丁-3-烯-2-醇(83mmol,99%,1.0mol当量),并加热至回流4h,在此期间无色溶液缓慢地变黑。然后将反应混合物冷却并倒入1200g冰水中,并通过小心地小批量先后添加(放热,放出二氧化碳)240g碳酸钠(2.25mol,工业级)和112g碳酸氢钠(1.27mol,工业级)中和至ph7。浅棕色混浊溶液用250ml乙酸乙酯萃取,然后用100ml乙酸乙酯萃取。合并的有机相用100ml盐水(10%nacl水溶液)洗涤,随后经硫酸钠干燥,过滤并在40℃/200-10mbar下蒸发。将不均匀的粗料(31.5g)在50ml庚烷/乙酸乙酯(90/10w/w)中消化并过滤。用50ml庚烷/乙酸乙酯(90/10w/w)洗涤滤饼(主要为hq)。将滤液真空浓缩(40℃/200-10mbar),得到粗产物(14.4g)。将该材料通过柱色谱法纯化;洗脱梯度庚烷至庚烷/etoac80:20(w/w)。合并纯级分并真空浓缩(40℃/200-10mbar)。将残余物溶于30ml二氯甲烷中,随后再次蒸发至干(40℃/200-0.1mbar),得到5.6g呈灰白色晶体的dmc(31.4mmol,通过qnmr的纯度为98.5%,产率37%)。
根据本发明的实施例:实施例2-13
实施例2:从1,4-氢醌和2-甲基丁-3-烯-2-醇开始合成2,2-二甲基苯并吡喃-2-醇
在装有氩气入口、磁力搅拌器、油浴和温度计的200ml4颈磺化烧瓶中装入35.1g氢醌(319mmol,99.5%,2.0mol当量),将其悬浮在77.7g硫酸(0.4m,0.17摩尔当量)和43.5克甲苯(50毫升)中。将两相反应混合物加热至回流(内部温度为85℃),之后所有氢醌都溶解在水相中。然后,在2小时内将14.0g的2-甲基-3-丁烯-2-醇(“mbe”)(159mmol,98.0%,1.0mol摩尔当量)添加至回流的反应混合物中。加完后,将反应在85℃(内部温度)下再搅拌3小时。在仍温热的同时,将反应混合物然后转移至分液漏斗,并分离无色相。保留水相(含有硫酸和过量的hq)用于下一个反应循环。有机相用去离子水(2×25ml)洗涤。然后将水相用甲苯(总共45g的甲苯(2x26ml))反萃取。然后将甲苯溶液在旋转蒸发仪中浓缩,以提供29.60g米色(beige)油状的粗dmc(经qlc测定为75.4%,相对于mbe的化学产率为78.7%)。在配备有vigreux柱(20cm)的蒸馏设备中,于0.3毫巴和125℃(内部温度)下纯化粗料,得到一馏分:22.40gdmc(通过定量lc为97.0%,121.9mmol,分离产率76.5%),mp.74.5-75℃。
实施例3:从1,4-氢醌和2-甲基丁-3-烯-2-醇开始合成2,2-二甲基苯并吡喃-6-醇:催化剂和1,4-氢醌相的再循环
在装有氩气入口、磁力搅拌器、油浴和温度计的1.5升4颈磺化烧瓶中,装入来自前次运行的651g水相(按与本次运行相同的规模进行;含氢醌,约3mol当量,~1.17摩尔,和硫酸,~0.4m,0.78摩尔,0.5摩尔当量,和~3摩尔%产物),以及另外的新鲜氢醌(43.0克,390mmol,99.5%,1.0摩尔当量);和溶于甲苯(500ml)中的2-甲基-3-丁烯-2-醇(33.7g,387mmol,99.0%,1.0mol当量)。将两相反应混合物加热至回流(内部温度85℃)4h。在仍温热的同时,将反应混合物然后转移至分液漏斗,并分离无色相。保留含有硫酸和过量hq的水相(约650g)用于下一个反应循环。用水(2×250ml)洗涤有机相。然后将水相用甲苯(250ml)反萃取。然后将甲苯溶液在旋转蒸发仪中浓缩,以得到米色油状的粗品2,2-二甲基苯并吡喃-6-醇(93.88g,通过定量lc的纯度为64.8%,产率为88%)。
实施例4:从1,4-氢醌和异戊烯醇开始合成2,2-二甲基苯并吡喃-2-醇(见图5)
在装有氩气入口、磁力搅拌器、油浴和温度计的350毫升4颈磺化烧瓶中装入17.5克1,4-氢醌(159mmol,99.5%,4.0mol当量)和异戊烯醇(3.45g,39.7mmol,99%,1.0mol当量)),然后将其悬浮于50ml硫酸(0.4m,0.5mol当量)和43.5g甲苯(50ml)中。将两相反应混合物加热至回流(内部温度84℃)4.5h。在仍温热的同时,将反应混合物然后转移至分液漏斗,并分离无色相。用甲苯(2×25ml)萃取温热的水相,并用水(2×25ml)洗涤合并的有机相,用na2so4干燥,并在真空中浓缩(40℃/50-20mbar),得到米色油状粗品dmc(7.0g,定量lc的纯度为76.3%,产率为76%)。
实施例5:由1,4-氢醌和异戊二烯开始的2,2-二甲基苯并吡喃-6-醇的合成(见图3)
在装有氩气入口、磁力搅拌器,油浴和温度计的200ml4颈磺化烧瓶中装入17.8g1,4-氢醌(161mmol,99.5%,4.0mol当量)和异戊二烯(2.75g,40.4mmol,1.0mol当量),然后将其悬浮在50ml的硫酸(0.4m,0.5mol当量)和43.5g的甲苯(50ml)中。将两相反应混合物加热至回流(回流期间内部温度缓慢升至82℃;在100℃下油浴)持续26小时。在仍温热的同时,将反应混合物然后转移至分液漏斗,并分离无色相。用甲苯(2×25ml)萃取温热的水相,并用水(2×25ml)洗涤合并的有机相,用na2so4干燥,并在真空中浓缩(40℃/50-20mbar),得到米色油状粗品dmc(5.7g,定量lc的纯度为80.5%,产率为64%)。
实施例6:在amberlyst15作为酸催化剂的存在下,由1,4-氢醌和2-甲基丁-3-烯-2-醇开始合成2,2-二甲基苯并吡喃-6-醇
在装有氩气入口、磁力搅拌器、油浴和温度计的200毫升4颈磺化烧瓶中装入8.0克1,4-氢醌(72.2mmol,99.5%,1.9摩尔当量)和2-甲基-3-丁烯-2-醇(4.0ml,39mmol,1.0mol当量),然后将其悬浮在50ml水和43.5g甲苯(50ml)中。加入amberlyst15(4.0g),并将两相反应混合物加热至回流(内部温度83℃)24小时。在仍温热的同时,将反应混合物然后转移至分液漏斗,并分离无色相。用甲苯(2×25ml)萃取温热的水相,并用水(2×25ml)洗涤合并的有机相,用na2so4干燥,并在真空中浓缩(40℃/50-20mbar),得到米色油状粗品dmc(5.8g,定量lc的纯度为80.6%,产率为67%)。
实施例7-9:
将与实施例5相同的反应规模和条件应用于其他催化剂。结果如下:
“molequiv.”=摩尔当量;“conc.”=浓度;“aq.”=水;“quant.”=定量;“lc”=液相色谱。
实施例10-13:浓度和mbe剂量的优化
为了降低hq摩尔当量以及所需的甲苯和硫酸的量,在2小时时间段内添加mbe,从而使hq/mbe的比例最大化。结果,可以通过将溶剂量减少到1/3并将每个循环中存在的hq量减少到1/2来提高时空产率。产率仅仅略微降低(从83%降至79%)。
表1.过量的hq、硫酸浓度和反应体积之间的折衷方案
*为两个阶段计算的mbe总浓度
水相中产物的量通常等于1-3mol%的产率。
比较例:实施例14:由1,4-氢醌(“hq”)和2-甲基丁-3-烯-2-醇(“mbe”)根据美国专利4,217,285中公开的条件,即在zncl2、二氧化硅-氧化铝和浓盐酸的存在下开始合成2,2-二甲基苯并吡喃-2-醇(“dm-苯并吡喃醇”)
“eq.”=摩尔当量,h=小时。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种基于二溴1,4?二丙基?1,4?二氮杂二环[2.2.2]辛烷的二价锰荧光材料及其制备方法 ...的制作方法
- 一种苯并二氮杂卓类化合物作为农用杀虫剂的新用图
- 一种苯并二氮杂卓类化合物作为杀菌剂的用图
- 一种(1s,4s)?2,5?二氮杂二环[2.2.2]辛烷及其衍生物的合成方法
- 包含噻吩并三唑并二氮杂卓化合物的药物制剂的制作方法
- 一种苯并内酯类化合物、其制备方法和在制备抗癌药物中的应用
- 二氮杂-苯并荧蒽并烯类化合物的制作方法
- 制备(2s,5r)-1,6-二氮杂-二环[3.2.1]辛烷-2-腈-7-氧代-6-(磺氧基)-单钠盐的方法
- 利用含有噻吩并三唑并二氮杂*化合物的药物制剂治疗白血病的方法
- 一种注射用短效苯并二氮杂卓盐药物组合物及其制备方法
- 2,4-O-二-α-L-吡喃鼠李糖基-β-D-吡喃葡萄糖基薯蓣皂甙的合成方法
- 一类6-芳基-3-环氨基甲基吡喃酮类衍生物的制备及用途的制作方法
- 吡喃葡糖氧基吡唑衍生物及其在药物中的应用的制作方法
- 取代亚甲基吡喃酮类衍生物及其制备方法和用途的制作方法
- 6-芳基-3-取代亚甲基吡喃酮类化合物及其制备方法和用途的制作方法
- 吡喃葡糖氧基吡唑衍生物、含该衍生物的药物组合物及其制备中的中间体的制作方法
- 7-糖氧基苯并吡喃衍生物及含有该衍生物作为活性成分的抗变应性剂的制作方法
- 取代的氨甲基苯并二氢吡喃在运动障碍和治疗锥体束外运动障碍药物引发的不利影响的 ...的制作方法
- 氨甲基取代的2,3-二氢吡喃并[2,3-b]吡啶类,制备方法和在医药中的应用的制作方法
- 3-甲基-八氢环庚三烯并吡喃-2-酮及其制备方法