用于在废水处理中脱除氮的方法和装置与流程
本申请是分案申请,其原申请的国际申请号为pct/us2013/059775,国际申请日是2013年09月13日,中国国家申请号为201380058150.1,进入中国的日期为2015年05月06日,发明名称为“用于在废水处理中脱除氮的方法和装置”。
相关申请的交叉引用
本申请要求于2012年9月13日提交的号为61/700,717的美国临时申请、于2012年10月1日提交的号61/708,498的美国临时申请和于2013年3月14日提交的号为61/783,232的美国临时申请的权益。号为61/700,717,61/708,498和61/783,232的美国临时申请通过引用并入本文。
背景技术:
本公开通常涉及废水处理系统和方法。通常而言脱除氮是一项处理任务,其在反应器体积、曝气能量和化学品用量方面需要最多的资源。本公开的目标在于在脱除氮中的更加节约资源的代谢途径-所谓的短程生物脱除氮。本公开的各个方面使得能够从废水中脱除氮以便以减少的能量、化学品和成本生产高品质的流出物。
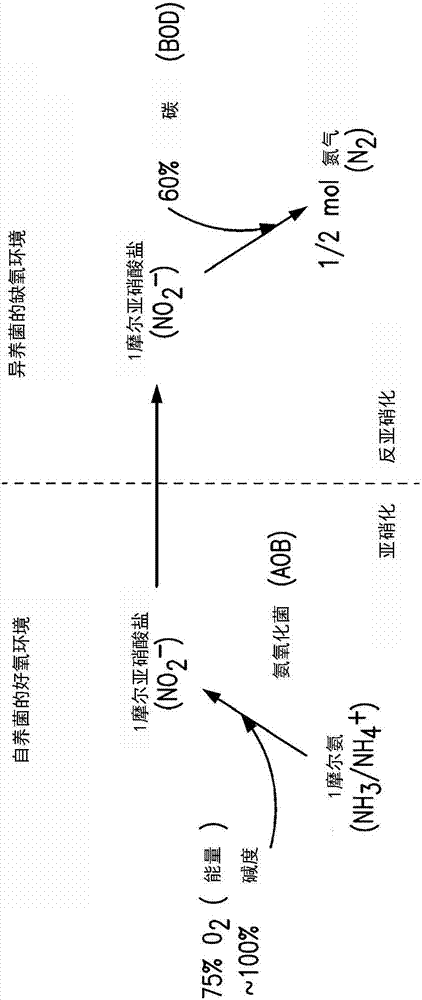
抑制亚硝酸盐氧化菌(nob)是用于实施短程生物脱除氮(scbnr)工艺的先决条件,所述工艺诸如为亚硝化-反亚硝化(nitritation-denitritation)(参见ciudad等人,2005年;gee和kim,2004年;ju等人,2007年;yoo等人,1999年;yu等人,2000年;zeng等人,2008年);亚硝酸盐分流(nitrite-shunt)和部分亚硝化-厌氧氨氧化(厌氧氨氧化(anammox))(参见fux等人,2002年;hippen等人,1997年;vandongen等人,2001年;wett,2006年;wett,2007年;wett等人,2010年);以及反氨化(deammonification)。通过控制nob而成功地抑制亚硝酸盐氧化相比于传统的硝化-反硝化(参见turk和mavinic,1986年;abeling和seyfried,1992年)节省25%的氧气和40%的有机碳。在反氨化工艺中,nob的控制产生在进一步降低所需的曝气能量、以及降低电子供体和固体处理成本方面的额外益处。图1、图2和图3示出用于分别通过传统的硝化/反硝化、亚硝化/反亚硝化和反氨化(部分亚硝化+厌氧氨氧化)来脱除氮的流程图。
相比于传统系统,由于不需要单独的罐以及将混合的液体(liquor)硝酸盐从好氧硝化区循环到厌氧反硝化区,因此特别希望在单个罐内进行同步硝化和反硝化(snd)。snd的益处通过利用亚硝酸盐分流路径而进一步扩展,如通过使用曝气持续时间的控制所证实的那样,所述控制利用氧化还原电位(orp)(参见guo等人,2009年)和氨ph曲线(参见peng等人,2004年)。反应器微环境(由于较差的混合和反应器设计的组合在反应器内形成的好氧和缺氧区)和絮体微环境(在活性污泥絮体内形成的好氧和缺氧区域)已被假定为适于snd的可能机制(参见daigger等人,2007年)。但是难于将控制策略并入到上述机制中以实现稳定的snd性能。据报道在分段的闭环反应器(诸如氧化沟,orbal)内发生snd(参见daigger和littenton,2000年),其通常采用较长的水力停留时间(hrt)、固体停留时间(srt)、和连续的低溶解的氧(do)。
鉴于为了满足日益严格的排放标准的高成本的生物营养物去除(bnr),通过抑制nob进行的scbnr是令人感兴趣的主题。在许多出版物中论述了为了了解nob抑制的尝试,其中包括更具体使用高温(参见hellinga等人,1998年,其公开内容以其整体明确地通过引用并入本文)、高含量游离氨抑制(inhibition)或do浓度(参见blackburne等人,2008年)和瞬时缺氧(参见kornaros和dokianakis,2010年)的那些。具体地,在各种方法中所有的这些条件部分地或作为整体使用,在处理高氨浓度废物流的系统中成功地控制nob,高氨浓度废物流诸如为厌氧消化池脱水液体(也通常在高温下)和填埋场渗滤液。高氨浓度的废物流在此限定为具有流入的进料,所述流入的进料具有大于200毫克/升的以氮计的氨浓度。在低浓度的废物流(诸如生活废水)中控制nob抑制仍然是一项挑战,并且是本公开的主题。下文描述在scbnr工艺中目前用于抑制nob的控制。
温度和氨:温度和游离氨两者是据信给氨氧化细菌(aob)提供优于nob的优势的特征。自从其由anthonisen等人考虑到(1976年)开始,nob的游离氨(fa)抑制已在文献中被充分证明,其公开内容以其全文通过引用明确地并入本文。然而,为了获得稳定的亚硝化而控制fa抑制的知识更有限,其原因在于nob适用性已经被报道(参见turk和mavinic,1989年;以及wong-chong和loehr,1978年)。此外,aob与nob相比,已知高温更有利于aob的生长(参见kim等人,2008年)。
在更高温度下相比于nob的aob的增加的活性,总氨更大解离成游离氨以及在更高温度下所得的nob抑制与低do操作结合(通常使用间歇曝气并用管理的好氧固体停留时间(srt)来进行)导致富集aob以及选择性去除(washout)nob。这些方法都不同地描述成在高氨浓度的废水中控制nob(参见ep0826639a1、ep0872451b1、us2010/0233777a1、us7846334b2、us6485646b1、和wo2012/052443a1)。所述方法使用悬浮生长(参见wo2006/129132a1)、在支撑介质上的附着生长(参见us2011/0253625a1和ep0931768b1)或颗粒污泥(参见wett,2007年;和us7846334b2)来完成scbnr。
尽管用来增加aob的活性以及用于控制nob的生长的升高的温度的作用是有效的,但是该作用在宽泛的温度范围下操作的低浓度的主流工艺中是不可行的。因此,在低浓度废水中的nob控制依然难于处理,并且需要小心操控除了温度或游离氨之外的因素。
溶解的氧(do):do在低氨浓度的废水中的nob控制上能起到显著作用。使用低溶解氧浓度的持续亚硝化已经在各种反应器配置中被观察到(参见sliekers等人,2005年;wyffels等人,2004年;和blackburne等人,2008年)。虽然这些报道缺乏基本机制的说明,但他们采取相比于nob的aob的较高氧亲合力的假说(参见hanaki等人,1990年;laanbroek和gerards,1993年;和bernet等人,2001年)作为针对所观察到的现象的解释(参见yoo等人,1999年;peng等人,2007年;lemaire等人,2008年;gao等人,2009年;和zeng等人,2009年)。sin等人(2008年)记载一种普遍的看法,即aob的氧亲和力大于nob的氧亲和力,以及低do的操作相比于nob更有利于aob,但是也有研究进行相反的报道(参见daebel等人,2007年;和manser等人,2005年),以及本发明人提出表明nob相比于aob的对低do-浓度的更强适应性的数据(参见图5至图6)。
生物强化和生物质富集:已经报道了从高浓度(strength)反应器到低浓度主流反应器的硝化生物质转移,因此用于执行硝化所需的srt在主流工艺中降低(us7404897b2,us6602417b1)。这种生物强化可从单独的侧流反应器或与主流反应器共连的反应器发生(parker和wanner,2007年)。还存在与下述相关的现有技术,即通过使用水力旋流器将主要含有厌氧氨氧化菌生物体的更密集性生物质部分物理分离并将该较重部分再循环以便富集非常缓慢地生长的生物质(ep2163524a1,us2011/0198284a1)。
瞬时缺氧:使用瞬时缺氧是用于实现nob抑制的常用方法(参见li等,2012年;ling,2009年;pollice等人,2002年;zekker等人,2012年;us7846334b2,ep0872451b1,和wo2006/129132al)。瞬时缺氧对于nob而言可引入滞后时间以便从缺氧过渡到好氧环境。kornaros和dokianakis(2010年)表明在瞬时缺氧之后的好氧条件下的nob恢复(recovery)上的延迟和nob生长的滞后,从而认可许多其他人的对瞬时缺氧的有用性的言论(参见allenman和irvine,1980年;katsogiannis等人,2003年;seldak,199;silverstein和schroeder,1983年;yang和yang,2011年;和yoo等,1999年)。虽然在由高游离氨抑制支持的“高浓度”废物中,瞬时缺氧已被成功地用于控制nob(参见wett,2007年;和us7846334b2),并已经提出在低浓度废物中使用瞬时缺氧的能力(参见peng等人,2004年),但在没有来自游离氨抑制的支持的情况下控制与瞬时缺氧相关联的特征的能力仍然是个谜。概括而言,用于在低浓度废水中控制nob选择出(out-selection)的策略不能产生可靠的工艺性能,其原因在于对最大化的aob速率尤其在nob的活性抑制不可行时成为nob选择出的关键缺乏了解,所述策略是新兴的scbnr技术的基础。在该背景下,我们限定术语“选择出”作为通过实现aob的更高速率而抑制nob的方法,从而产生针对其它生物体的亚硝酸盐。
技术实现要素:
本公开使用各种控制策略实现维持高的aob氧化速率的合适和可测量的控制的同时,实现nob选择出,所述策略包括:1)曝气控制,其确保接近饱和的氨浓度和do分布(profiles),所述do分布从高的设定值到迅速降低接近于零来改变,用于在好氧阶段中的最大化的aob速率;2)创新性的曝气控制策略,其基于在线测得的nh4-和nox信号比以便将总氮的脱除最大化,并提供用于厌氧氨氧化反应的最佳化学计量的底物(substrate)配比;3)用于去除nob上的压力的积极的好氧srt管理;以及4)厌氧氨氧化菌和更轻的絮体(flocculant)aob部分的生物强化。使用这样的控制策略提供优于现有技术策略的重要优势。
因此,本公开提供在用于生物脱除氮的反应器中从废水脱除氮的系统和方法,其中在反应器中的好氧-缺氧的持续时间和/或溶解氧的浓度基于实时测得的氨浓度与氮氧化物(oxidizednitrogen)(nox,亚硝酸盐和硝酸盐浓度的总和)的比值来控制。通常而言,在反应器中的好氧-缺氧的持续时间和/或溶解氧的浓度经受控制以达到处于0.5至1.5范围内、优选在0.7至1.0之间的氨浓度与氮氧化物的浓度的比值。通过采用本公开的系统和方法,总体脱除氮被最大化,因为反硝化(取决于化学需氧量(cod)的输入)和随后的氨氧化彼此平衡,同时相比于nob还更有利于aob。替代性地,代替应用比值,氨浓度和氮氧化物浓度之间的差可被控制为接近0的值,其导致等效的控制性能。
本公开可用于实现主流snd工艺的合适和可测量的控制,其通过几个脱除氮机制中的一个将总无机氮(tin)的脱除最大化,所述机制包括硝化-反硝化、亚硝化-反亚硝化(scbnr)、通过在单独的下游反应器中的厌氧氨氧化而产生适用于精制(polishing)的流出物流的部分亚硝化-反亚硝化,和在具有选择性的厌氧氨氧化菌保留性的单个罐内的部分亚硝化-厌氧氨氧化。这些系统和方法使用下述控制策略,所述策略包括:1)氨、亚硝酸盐和硝酸盐的实时测量;2)基于在反应器中测得的氨浓度与氮氧化物浓度的比值所控制的操作性的do和do设定值的正确使用;3)曝气频率的控制,所述控制基于在反应器中测得的氨浓度与氮氧化物浓度的比值;以及4)在宽泛范围的装置(反应器配置)和操作条件下的瞬时缺氧的正确实施。
附图说明
图1是示出与传统硝化和反硝化相关联的反应的摩尔流程图。
图2是示出与亚硝化和反亚硝化相关联的反应的摩尔流程图。
图3是示出与反氨化相关联的反应的摩尔流程图。
图4是在blueplainswastewatertreatmentplant(wwtp)(左)处的实验室规模的间歇式反应器中和在strasswwtp(右)处的全规模中的总硝化菌(aob+nob)和仅nob的ko-值的所收集数据的比较的线图。
图5是示出具体氮工艺速率的绘图测试数据的曲线图(左),其根据取决于在blueplains(wwtp)处的间歇式反应器的间歇曝气的do-设定值的每克挥发性悬浮固体(vss)每天所脱除的氨;通过应用最小平方误差最小化而将monod表达式(右)拟合到测得的数据(箭头指示在1.5毫克/升的do水平下比aob的n-处理速率高15%)。
图6是在hamptonroadssanitationdistrict(hrsd)(wwtp)处的亚硝化反应器中的aob和nob的ko-值的所收集数据的比较的线图。
图7是用于实施本公开的系统的示意图。
[保留]
[保留]
[保留]
[保留]
[保留]
[保留]
图8是在用相同的曝气方案操作的两个相同序批式反应器sbr之间的do消耗曲线的比较的时间曲线图,其中一个被供以初级处理的流出物(pe)以及另一个被供以二次处理过的流出物(se)。注意在pe供应系统中较高的化学需氧量(cod)允许从高的do设定值更迅速地过渡到缺氧。
图9是在用间歇式曝气[剩余氨]操作的sbr配置中在反应阶段期间的氮种类(nh3、no2和no3)的曲线的比较的图。
图10是在用间歇式曝气[无残留的氨]操作的sbr配置中在反应阶段期间的氮种类(nh3、no2和no3)的曲线的比较的图。
图11是装配有机械搅拌器、空气扩散器、氨、亚硝酸盐、和硝酸盐的bnr反应器和do分析仪的侧向横截面视图。
图12是示出氨相对于nox(avn)和氨控制的曲线图,所述氨相对于nox(avn)和氨控制利用(nh4+-n-nox-n)和nh4+-n的波动而调节好氧和缺氧的持续时间。
图13是曲线图,其示出通过比较aob和nob速率选择出nob的程度以及在hrsdwwtp处的亚硝酸盐分流反应器中的得到的亚硝酸盐的积累。
图14是通过定量的聚合酶链反应(qpcr)的测量而比较nob速率和nob群落的曲线图。
图15描绘主流反氨化控制器(pilot),其具有亚硝酸盐分流的连续搅拌罐式反应器(cstr)随后是厌氧氨氧化移动床生物反应器(mbbr)。
图16呈现流入的氨和亚硝酸盐以及来自厌氧氨氧化mbbr的流出的氨和亚硝酸盐。
具体实施方式
本公开提出用于通过氨、do、生物强化、和瞬时缺氧控制(nicita)来提高aob氧化速率以及选择出和控制nob的方法。下面描述与这四个特征相关联的本公开和用于实施本公开的控制。本公开涉及用于将氮从在反应器中处理的废水中脱除的系统和方法。本公开的系统和方法通过瞬时缺氧和好氧srt的控制、选择出nob、以及通过保持氨(nh4)浓度与氮氧化物浓度的预定比值来控制动态do浓度或曝气间隔而将氮的脱除最大化,同时将曝气和有机碳的要求最小化。氨浓度与氮氧化物浓度的优选比值可在0.7到1.0之间,但该比值可大至0.5到1.5之间。利用这些动态控制策略的控制器已被命名为avn(nh4相对于nox)。avn控制不仅将通过正常途径(图1)而将脱除tin的潜力最大化,而且它还提供选择出nob的机会,以及在根据图2和图3的脱除tin方面的相关联益处。附加的控制目标是提供两种底物(氨和亚硝酸盐)的接近为0.76的实际化学计量比nh4-n/nox-n的合适混合物。代替比值,替代性地[氨(nh4)浓度]和[亚硝酸盐和硝酸盐浓度的nox-n的总和]的负值的总和可在目标在于-3和+1之间的范围内的控制算法中使用。这些目标值反映下述事实,即通常而言氨对接收到的水体是更毒的,且因此厌氧氨氧化反应的化学计量比也需要比氨多32%的亚硝酸盐。
氨:本公开的主要建议偏离更通常使用的温度和游离氨来实现选择出nob。而使用温度来控制aob和nob的相对生长速率在主流工艺内并不总是可能的,根据本公开,可采用控制特征,其将氨本身的直接测量值用作为控制变量来代替游离氨和温度。本公开使用氨氧化程度的直接控制,使得氨在整个指定的反应器时间周期或特定的反应器长度内被氧化。在整个反应器长度或时间周期内允许保持残留的氨将保持nob上的压力,同时保持高的aob速率。
本公开利用在bnr反应器中的氨、亚硝酸盐、硝酸盐和do的直接测量,以控制好氧和缺氧的srt和hrt以及反应器do浓度以便将氨氧化和反硝化最大化。溶解氧的浓度或曝气间隔或两者取决于流入的碳:氮(c/n)的比值和反应器条件来进行有效地控制,使得在任何给定时间下有利于用于脱除氮所需的反应。do相比于cod氧化更多地是针对氨氧化,以及可用的cod用于在所有的时候驱动反硝化,因此,将总氮脱除最大化。由本公开允许的氨氧化的程度由用于反硝化所引入cod的可用性来控制,因此本质上氨氧化和反硝化通过彼此平衡以便将氮的脱除最大化。do浓度和/或曝气持续时间通常受到控制,以便在反应器中在任何时候都维持大致相等的nh4-n和nox-n浓度,nh4的氧化量以及从而所输送的氧气量基于可用于将所产生的nox脱氮的引入cod的量来控制。这将好氧异养的cod消耗最小化,以及将反硝化的机会最大化,这在低的do和可用的cod下需要时间。控制器允许输入偏移(offsets),其将允许所述nh4-n或nox-n浓度被脱除,以满足针对这些参数的特定排放限制。例如,控制器可被调谐,以确保通过设置控制器来符合nh4极限值,以便在20-90%的流出物nox-n浓度下提供包括nh4的流出物。
溶解氧:虽然上面所述的论文中提出亲和力上的差异(例如,相比于aob,nob的对于低溶解氧浓度的更强的适应),由本发明人发现在控制方案中采用这些差异的机会。因此,令人惊讶地且违背于先前的解释,瞬时高的do操作显现出更适于竞争出(out-compete)nob。使用较高do水平的情况是本公开与现有技术的差异。较高的do(>1.0毫克/升)不仅保持较高的aob速率,而且还控制使aob和nob的相对底物亲和力处于朝向nob选择出的状态。
如上文所述以及在图4至图6中针对三种不同的设置(plant)构造所示,在高的do浓度(即,浓度大于1毫克/升)下相比于亚硝酸盐氧化,以更快的速率发生氨氧化。因此,希望在瞬时地高do浓度下操作bnr反应器,使得相比于nob有利于aob的生长。该策略与大量的文献相反,其指示相比于nob的aob的高的氧亲和力,以及用来选择出nob的在低溶解氧浓度下的优选的操作。
生物强化:不同于现有技术的方法,现有技术方法的目的在于从高浓度的工艺将整个生物质进行生物强化,而本公开的目的在于选择主要包含aob的较轻的生物质部分(即,溢流而不是水力旋流器的底流),以便将从侧流反应器到主流的相对快速生长的aob进行生物强化,而在经受反氨化的侧流反应器中不会以不受控的方式损失厌氧氨氧化的活性。使用旋风分流器(cyclone)、或筛选择更轻或更细的生物质部分或者从生物膜载体介质分离的未附着生物质允许最大的播种量(seedingrate),这有助于在高浓度反应器(选择性地降低srt)和低浓度的系统(传送aob,但几乎不传送nob)两者中抑制nob。以类似的方式,对来自侧流或高浓度反应器的厌氧氨氧化生物体的生物强化也是可能的。本公开给主流bnr反应器a提供厌氧氨氧化菌的生物强化(图7),其中do保持>1.0毫克/升,以抑制nob。在最佳条件下的生物强化的生物体被保留,并允许在bnr反应器a中生长。
图7示出用于从废水102脱除氮的系统100。系统100包括反应器a,其用于对废水102执行生物脱除氮。分析仪b、c都位于反应器a中。第一分析仪b感测反应器a中的氨,并产生相应的氨信号12,以氮计的方式表示。第二分析仪c感测在反应器a中的亚硝酸盐、硝酸盐、或亚硝酸盐和硝酸盐的组合,并产生相应的亚硝酸盐、硝酸盐、或亚硝酸盐和硝酸盐的组合信号14,都以氮计的方式表示。控制器d接收信号12、14,并且使用信号12、14来产生对气流控制装置e的指令信号16。装置e在指令信号16的控制下操作,以调节空气的流动或供给到反应器a的曝气104时间。因而,被供给到反应器a的空气104的量基于从控制器d所接收到的控制信号16。来自反应器a的流出物106被供给到第二反应器f。流出物106适于进一步脱除剩余的氮,并且因此由第二反应器f进行氮的进一步脱除。
瞬时缺氧:本公开还提供成用于迅速过渡到缺氧或从缺氧的迅速过渡。用于从高do迅速过渡到缺氧的情况是本公开的另一个主题。快速过渡到缺氧不仅允许对于nob生长而言的很少的机会,而且从缺氧迅速过渡回到高do设定值还允许抑制nob的生长。从缺氧的快速过渡还可产生抑制nob的中间体。从本质上而言亚硝酸盐氧化在氨氧化之后。大多数模型使用基于monod方程式(monod,1949年)的底物半饱和来描述使用电子给体和受体的能力。在快速变化的do环境中,某些亚硝酸盐可能基于与nob相关联的底物半饱和而朝向氨氧化的结束(end)积聚。在这种情况下,如果曝气继续,则残留的亚硝酸盐最终将通过nob转化为硝酸盐;然而,如果曝气被中断以及条件是允许快速过渡到缺氧状态,则残留的亚硝酸盐将通过在亚硝酸盐分流工艺中由异养反硝化菌驱动的化学需氧量(cod)或通过在单段反氨化工艺中的厌氧氨氧化以“非氧化(anoxically)”的方式减少。因此,亚硝酸盐还原菌(例如,异养反硝化菌或厌氧氨氧化菌)可竞争过nob(通过消耗亚硝酸盐),从而通过限制它们底物的可用性而在nob上形成压力。nob群落将在每一个后续周期内获得越来越少的能量,进一步降低其有活性的群落。因此,关键的是在氨氧化结束时要限制曝气并迅速地过渡到缺氧,使得当亚硝酸盐可用时剥夺nob的do。
根据本公开,通过增加混合液的固体浓度(约2克/升[在1.5克/升和4.0克/升之间)或通过在缺氧阶段引入化学需氧量(c/n>2)以便迅速清除氧来维持较高的氧吸收速率(10分钟内实现缺氧)而允许迅速过渡到缺氧。另一种方法是增加温度(例如,使用来自涡轮机或发动机的废热),并因此增加所有生物体的生长速率。图8示出对于以间歇式曝气方式操作的两个sbr系统而言的do分布的比较的这样的实例,其中一个被供以初级处理的流出物以及另一个被供以二次处理过的流出物。根据该策略,nh4-n被测量并保持在某些预定的nh4-n设定值范围内或在具有合适带宽或等于反应器的nox-n浓度的单一设定值,这取决于反应器的水力工况。这种方法因为始终保持残留的氨消除了在接近完成氨氧化之后的过度曝气的担忧,这进一步有助于保持较高的aob速率。因此,根据本公开能够采用已知的选择出nob的策略,所述策略利用基于直接的氨、亚硝酸盐和硝酸盐的鲁棒控制算法和do信号。从缺氧快速过渡到曝气状态通过使用适当的曝气设备来实现,所述曝气设备可迅速地供应空气或通过连续的好氧和缺氧区。相对于在曝气阶段期间在给定的do设定值下采用稳定的氧气吸收所需的曝气容量,在该背景下的适当的曝气设备是通过鼓风机和扩散器的氧气供应的额外容量。该额外容量(超出基线设计约10-25%)需要使得do-增加从较低设定值(<0.1毫克/升)加速增加到上限设定值(>1.0毫克/升)。图9和图13示出在控制nob时的这种策略的性能,以便分别实现在亚硝酸盐分流系统和单段反氨化系统中的scbnr。图10示出过度曝气(overaerating)对nob的选择出(由产生硝酸盐指示)的负面影响(由无残留的氨表示)。
用于通过这四个可能的特征(氨、do、生物强化和瞬时缺氧控制)控制nob的特定控制描述如下:
好氧srt和do设定值:如切实可行的那样,理想的是保持do设定值为1毫克/升以上。较高的do设定值允许相比于nob的aob的更迅速生长。然而,非常高的do设定值增加过渡到缺氧所需的时间。已成功地用于实施这种高do策略的一种方法在下面的控制叙述中进行描述。应当注意的是,这种高do策略违背于针对低浓度实现亚硝酸盐分流的传统观念以及在高浓度和高温废气流中实现亚硝酸分流的实际实践。
好氧srt通过两种方法来控制。固体废物的增加降低了总的好氧srt。减少好氧srt的第二种方法是在瞬时缺氧过程中通过增加缺氧时间步骤来实现的。在avn和氨控制策略下操作的间歇式曝气(在时间或空间上)的bnr反应器中,好氧srt通过用于将氨氧化成亚硝酸盐的aob的曝气需求来确定,使得ⅰi)nh4-n浓度等于nox-n浓度或ii)nh4-n浓度等于设定值。例如,如果aob的氨氧化速率越低,则与aob速率较高的情况相比将要求更多的曝气(时间或更高的do浓度或两者)来保持该状态。在这种情况下,总体srt的有意降低逐渐导致在特定do值下的aob氨氧化速率的降低。因此,aob需要更多的曝气来增加它们的生长速率以及满足所需的条件(nh4-n=nox-n或设定值),导致操作性的高do设定值(在时间上)和好氧hrt(在空间上),以便增加到并处于相比于nob的生长的更有利于aob生长的值。
积极的srt控制未被普遍接受为实现亚硝酸盐分流的手段,这也与无法维持稳定的选择出nob相一致。当bnr反应器在高do设定值下操作时,aob比nob生长地更快,这允许系统在进一步不利于nob的低的srt下进行操作。此外,根据本公开容易控制积极的srt压力的施加。由于氨、亚硝酸盐和硝酸盐的浓度确定操作性的高do设定值或曝气的持续时间(在时间上)和曝气部分(aeratedfraction)(在空间上),本公开提供用于控制总srt的并不复杂的系统,以使得do保持在高浓度下,超过1毫克/升。
nob的外部和内部抑制剂或毒剂:通过将nob暴露于抑制剂或有毒的物质,例如但不限于游离氨、游离亚硝酸、和其它天然或合成的物质,来减少nob的净生长速率是可能的。还可以设想到利用内部(在污水处理厂内)产生的诸如一氧化氮、羟胺、肼等的含氮代谢产物或其它处理设备的中间体(诸如来自热水解或其它发酵过程)而根据需要通过控制这些中间体的生产来抑制nob。抑制剂也可被添加到工艺中以便降低nob的生长速率。
适于aob和厌氧氨氧化菌的生长增强剂:使用适于aob或厌氧氨氧化菌的生长增强剂也是可能的。例如,一种方法是使用肼作为适于厌氧氨氧化菌的生长增强剂,同时对于nob而言是毒剂。使用抑制剂或生长增强剂的总体目标是增加和区分aob或厌氧氨氧化菌相对于nob的相对生长速率以便最终选择出nob。
后缺氧脱除氮:来自bnr工艺a的含有氨、亚硝酸盐和硝酸盐的流出物(图7)可进一步在后缺氧工艺f中进行处理,后缺氧工艺f使用微生物,诸如但不限于增加有或没有增加有机电子供体的厌氧氨氧化菌。bnr反应器a和反应器f可以是集成的反应器(使用单个固液分离装置)或独立的反应器(在它们之间具有固-液分离装置)。完全缺氧反应器f配置成使用几种类型的工艺选择而选择性地保持厌氧氨氧化菌,所述工艺选择诸如为活性污泥后缺氧反应器、颗粒污泥反应器、移动床生物膜反应器、,集成的固定膜活性污泥反应器、具有生物活性的过滤器或膜生物反应器。用于这类脱除氮的有机外部电子供体可包括但不限定于有机碳源或无机源,所述有机碳源诸如为乙酸盐、乙酸、甲醇、乙醇、甘油、糖和它们的组合,所述无机源诸如为硫化物,元素硫,元素铁等。后缺氧也允许在反应器f内的厌氧氨氧化生物体产率上的增加,过量的这些生物体可以再循环至反应器a。
反应器配置:一些设备可用于执行该aob氧化和选择出nob的方案,所述设备包括完全混合的反应器(completemixedreactors),序批式反应器、氧化沟和塞流反应器。应当指出的是,反应器设备可被调节以便给予用于实现srt、氨氧化要求、高do设定和缺氧过渡的控制特征,其中在可能的情况下上述控制特征通过提供用于获得在溶解氧设置和空间上缺氧或在时间上缺氧的设置的机械和液压灵活性来实现。可以设置摆动区域(任选地(facultatively)曝气反应器体积)或反应器,其用于容纳对于废水处理过程是通常情况的可变流动和负载。除了悬浮生长反应器之外,生物膜、颗粒污泥或这些反应器的混合也是可行的。最后,可使用任何独立的装置(包括澄清器、膜或溶气气浮罐)来进行固-液分离。
塞流式反应器的特征在于其是连续地供给反应器,其具有非常高的长度与宽度的比,并且可模拟成一系列完全混合的反应器,其中污染物浓度沿着横跨(across)反应器长度的流动通路而减小(即浓度梯度)。在大型处理厂中是更为常用的的塞流式连续供给的反应器中,用于实现scbnr的处理控制可使用以下两种配置来解决:(1)通过在好氧和缺氧区域之间交替来在空间上控制曝气;和(2)通过以类似于sbr配置的方式使得空气以“送入空气(airon)”和“断开空气(airoff)”的顺序在整个反应器中循环来在时间上控制曝气。控制混合液和好氧/缺氧过渡所需的其它特征类似于上述的sbr方案。
控制策略:若干控制策略是可用的,所述策略可在上述的反应器配置中应用,所述配置使用本公开的特征以实现最大的tin脱除并可扩展到选择出nob。下面描述了一些示例性的策略,其针对不同的配置进行优化。
控制策略a(好氧–缺氧时间控制):在该策略中,do设定值是固定的,而好氧和缺氧时间是可变的。总体好氧-缺氧循环时间可保持在一定的设定值下,同时允许改变好氧和缺氧的持续时间。在另一实例中,缺氧的持续时间可以是固定的,从而使得所述控制器仅仅改变好氧的持续时间,这样使得取决于脱除氮的可能性,总体的缺氧加上好氧的持续时间保持是动态的。当未设置曝气时应设置机械混合。在该实例中,好氧和缺氧的持续时间可在16分钟循环时间内的4分钟和12分钟之间变化(图12)。当在反应器中的nh4-n大于nox-n(avn控制)和氨设定值(氨控制)时,好氧持续时间增加,直到nh4-n低于nox-n和设定值。当nh4-n浓度低于nox-n时,降低好氧持续时间,直到nh4-n浓度高于nox-n和氨设定值(图12)。当曝气的do维持在>1.5毫克/升时,支持与如上所述的亚硝酸盐氧化速率相比的更高的氨氧化速率。可如上所述应用偏移,以允许相对于nox的更多nh4流出物,或根据需要反之亦然。
在该示例性实施例中,bnr反应器20(图11)可装配有溶解氧、氨、亚硝酸盐和硝酸盐的传感器(或分析仪)22、24、26。将可能具有任何合适的反应器配置,其中控制可在时间或空间上发生。在多个(multiple)或塞流反应器的情况下,多个do探头沿着行列(train)的每个主要部段安装,而氨、亚硝酸盐和硝酸盐探头将战略性地安装在后面的反应器或部段中,以便控制反应速率,使得在反应器的端部处维持少量的氨浓度,并使得所述反应器流出物含有约等于nox-n(avn控制)或氨设定值(氨控制)的nh4-n浓度。
控制策略b(do强度控制):在该策略下,操作性的do是可变的,并由bnr反应器中的nh4-n和nox-n浓度控制,上述浓度将针对高氨氧化速率和在缺氧的异养反硝化或厌氧氨氧化驱动的氨氧化下优化do。这种方法在宽泛范围的反应器配置中是有效的,包括塞流式、完全混合、串联的完全混合反应器、以及序批式反应器。根据这种方法,do在低do设定值(其是固定的)和可变的高do设定值之间循环,所述高do设定值通常大于1毫克/升,并通过相比于设定值(氨控制)或nox-n浓度(avn控制)的反应器中的nh4-n来控制。维持积极的好氧srt以增加对氧的需求,从而允许控制器自动增加do水平至大于1毫克/升。在这种控制策略中,好氧和缺氧阶段由aob的曝气要求规定,以满足nh4-n邻近设定值(氨控制)或nox-n浓度(avn控制)的目标。
控制策略-c(摆动区域控制):氨和avn控制也可在塞流式流动或步进供给(在相继的缺氧区域内的几个供给点)的罐中使用,所述罐具有多个连续的串联的好氧和缺氧摆动区域。影响串联区域的控制(摆动区域可曝气或保持缺氧)保持缺氧或好氧来达到控制的目的。
控制策略的实例包括但不限于以下内容:
1.亚硝酸盐分流:用2-3小时的水力停留时间(hrt),~5天的srt,3500±500毫克/升的混合液悬浮固体(mlss),在25℃下操作用于脱除在反应器20中的氮的系统(图11),用于从本公开的废水生物脱除氮。反应器在图11-12中所示的avn控制策略下操作。图13代表观察到的选择出nob。图14示出优势nob(硝化螺旋菌)的趋势与nob速率测量值的趋势匹配,其清楚地证实观察到的选择出nob。
2.厌氧氨氧化抛光:图15代表来自上述实例的亚硝酸盐分流反应器,其后是用于氮精制(nitrogenpolishing)的完全未曝气且混合的厌氧氨氧化mbbr。反应器在4小时的hrt下操作,用50%k3(anoxkaldnes)培养基灌装。厌氧氨氧化mbbr表明几乎完全脱除no2-(厌氧氨氧化流出物no2--n=0.16±0.09毫克/升),同时nh4+脱除已经受到在亚硝酸盐分流流出物中的no2-积累水平的限制(图16)。
现在参照图1,用于实施本公开的系统200可包括生物脱除氮的反应器20、氨分析仪26、以及用于处理氨浓度信号30的控制器28,用于在受控的瞬时缺氧条件下实现选择出nob的目标。根据本公开,所述条件可沿着流动路径或者沿着处理时间线受控制。在操作中,反应器20的do分布在小于0.1毫克/升的较低do设定值和大于1.0毫克/升的较高do设定值之间切换,以及曝气系统32的激活间隔、较高的do设定值或两者都被设定,使得对于在空间或时间上高于瞬时曝气反应器体积的75%时,在线测量的氨浓度(30)高于1.5毫克/升。
图11的系统可另外包括分析仪24,其感测反应器20中的氮氧化物的浓度,并产生代表亚硝酸盐、硝酸盐或亚硝酸盐和硝酸盐的组合的浓度信号34。在操作中,控制器28处理针对氨和氮氧化物浓度的信号30、34,以便基于反应器20中的氨浓度和氮氧化物浓度的比值或总和来控制在反应器20中的(36)do浓度或好氧阶段的持续时间和/或缺氧阶段的持续时间。
控制器28、38可用于产生指令36、39,用于增加、降低或维持溶解氧的浓度(40)或好氧阶段的持续时间和/或缺氧阶段的持续时间,以维持从约0.5至1.5的氨浓度与氮氧化物浓度的比值,或用于维持在从-3.0至1.0内的氨浓度加上氮氧化物浓度的负值的总和。
根据本公开,反应器20可具有被限制的好氧污泥停留时间,其恰好足以通过缺氧阶段或体积的受控延展和通过操作高污泥废弃率而达到氨设定值42,使得在do强度控制的情况下污泥废弃率受到控制以维持大于1毫克/升的高do(40),或在好氧/缺氧持续时间控制的情况下通过减少缺氧的持续时间来增加好氧的持续时间,以便提供最小的好氧部分。
此外,系统200可包括由高氨浓度反应器52所产生的aob50的生物强化,所述高氨浓度反应器52被限定为在供给中具有超过200毫克/升的以氮计的氨,其中更小的密度的或更可压缩或未附着的污泥部分从高氨浓度反应器52选择,并以最大的速率供给(50)到反应器20,使得在高氨浓度的氨反应器52中小于10天的生物强化部分的保留时间,所述高氨浓度反应器52用于通过适当的装置54的生物强化,所述装置54诸如为旋风分流器、离心机、薄片式澄清器(lamellasettler)、丝网(screen)、或集成的生物膜反应器。
在操作中,控制器28接收氨浓度信号30和亚硝酸盐的浓度信号、硝酸盐的浓度信号或亚硝酸盐和硝酸盐的浓度信号的组合34,并产生指令36,用于基于在反应器20中的氨浓度与氮氧化物浓度的比值或总和来增加、降低或维持在反应器20中的do或好氧和缺氧的持续时间。
根据本公开,主要的设备氮脱除反应器20可包含来自高氨浓度的反应器52的厌氧氨氧化生物体的生物强化50,以提供有助于选择出nob的针对亚硝酸盐的微生物竞争体。
根据本公开,反应器20可维持氧吸收速率,其允许以少于10分钟的时间从曝气状态迅速过渡到反应器20中的缺氧状态,并使用适当的曝气装置32、56、58、60,其允许从缺氧状态迅速过渡到曝气状态。
再次参照图7,分析仪c可用于感测在反应器a中的氮氧化物的浓度。分析仪c产生浓度信号14,其可代表亚硝酸盐、硝酸盐或亚硝酸盐和硝酸盐的组合的浓度,并且控制器d处理分别针对氨浓度和氮氧化物浓度的信号12、14,并且基于氨浓度与氮氧化物浓度的比值或者总和来控制在反应器a中的(16)do浓度或好氧阶段的持续时间和/或缺氧阶段的持续时间。由受控工艺产生的流出物106被供给到完全缺氧的配置f中,其可选择性地生长和保留厌氧氨氧化菌。后一配置f可以是集成的或独立的后缺氧活性污泥反应器、颗粒污泥反应器、移动床生物膜反应器、集成的固定膜活性污泥反应器、具有生物活性的过滤器、或膜生物反应器,其可以将过量的生物质从这种反应器f再循环或生物强化到上游bnr反应器a。
在操作中,来自反应器a的流出物流106(图7)包含氨、亚硝酸盐和硝酸盐的混合物,使得可利用在完全缺氧悬浮或生物膜反应器f中的厌氧氨氧化进一步完成脱除氮,其中乙酸酯(acetate)、乙酸或某些其它有机或无机底物或底物的组合添加到反应器f中,通过厌氧氨氧化菌或其它微生物实现选择性地将硝酸盐还原成亚硝酸盐以及随后通过厌氧氨氧化菌实现将亚硝酸盐和氨脱除。
如果需要,nob的净观察到的生长速率可通过暴露于抑制剂、氮代谢中间体或有毒物质来降低,所述抑制剂、氮代谢中间体或有毒物质诸如为游离的亚硝酸、肼、游离氨、羟胺、和一氧化氮。此外,如果需要,aob和厌氧氨氧化菌的净生长速率可以使用生长因子诸如肼来增加。
上文参照的文献
美国专利文献:
us2010/0233777a19/2010chandran等人
us6485646b111/2002dijkman等人
us2011/0198284a18/2011nyhuis
us2011/0253625a110/2011takeda等人
us7846334b212/2010wett
外国专利文献:
ep0931768b19/2003
ep0826639a13/1998
ep0872451b112/2002
wo2006/129132a112/2006
wo2012/052443a14/2012
其它出版物:
abelingandseyfried(1992)waterscienceandtechnology26(5-6):1007-1015.
allemanandirvine(1980)waterresearch14:1483-1488.
anthonisenetal.(1976)journalofwaterpollutioncontrolfederation48:835-852.
bernetetal.(2001)journalofenvironmentalengineering127:266-271.
blackburneetal.(2008)biodegradation19(2):303-312.
ciudadetal.(2005)processbiochemistry40(5):1715-1719.
daebeletal.(2007)waterresearch41:1094-1102.
daiggerandlittleton(2000)waterenvironmentresearch72:330.
daiggeretal.(2007)waterenvironmentresearch79:375fuxetal.(2002)journalbiotechnology99(3):295-306.
gaoetal.(2009)bioresourcetechnology100:2298–2300.
geeandkim(2004)waterscienceandtechnology49(5-6):47-55.
guoetal.(2009)processbiochemistry44:979–985
hanakietal.(1990)waterresearch24:297-302.
hellingaetal.(1998)waterscienceandtechnology37(9):135-142.
hippenetal.(1997)waterscienceandtechnology35(10):111–120.
juetal.(2007)waterenvironmentresearch79(8):912–920.
katsogiannisetal.(2003)waterscienceandtechnology47(11):53–59.
kimetal.(2008)processbiochemistry43(2):154-160.
kornarosanddokianakis(2010)environmentalscienceandtechnology44:7245–7253.
laanbroekandgerards(1993)archivesofmicrobiology159:453–459.
lemaireetal.(2008)biotechnologyandbioengineering100(6):1228-1236.
lietal.(2012)processsafetyandenvironmentalprotection.doi:10.1016/j.psep.2012.05.009
ling(2009)proceedingsof2ndiwaspecializedconferenceonnutrientmanagementinwastewatertreatmentprocessespp.403–410.
manseretal.(2005)waterresearch39(19):4633-4642.
pengetal.(2004)waterscienceandtechnology50(10):35-43.
pengetal.(2007)chinesejournalofchemicalengineering15(1):115-121.
polliceetal.(2002)waterresearch36(10):2541–2546.
sedlak(1991)phosphorusandnitrogenremovalfrommunicipalwastewater:
principlesandpractice(2nded.).
silversteinandschroeder(1983)journalofwaterpollutioncontrolfederation55:377-384.
sinetal.(2008)waterscienceandtechnology58(6):1155-1171.
sliekersetal.(2005)appliedmicrobialbiotechnology68:808–817.
turkandmavinic(1989)waterresearch23:1383-1388.
turkandmavinic(1986)canadianjournalofcivilengineering13:600–605.
vandongenetal.(2001)waterscienceandtechnology44(1):153-160.
wett(2006)waterscienceandtechnology53(12):121–128.
wett(2007)waterscienceandtechnology56(7):81–88.
wett(2010)waterscienceandtechnology61(8):1915–1922.
wong-chongandloehr(1978)waterresearch12(8):605–609.
wyffelsetal.(2004)waterscienceandtechnology49(5-6):57-64.
yangandyang(2011)journalofhazardousmaterials195:318-323.
yooetal.(1999)waterresearch33:145–154.
yuetal.(2000)journalofenvironmentalengineering126:943-948.
zekkeretal.(2012)environmentaltechnology33(4-6):703–710.
zengetal.(2008)chemicalengineeringandtechnology31(4):582-587.
zengetal.(2009)enzymeandmicrobialtechnology45(3):22.
本发明并不局限于上述和附图所示的结构、方法和手段。本发明由下面所提出的权利要求来限定。所主张的权利和期望由美国专利证书保护的是下述权利要求。
- 还没有人留言评论。精彩留言会获得点赞!