一种金属元素晶格掺杂Si基材料催化剂的制备方法及甲烷无氧制乙烯的方法与流程
本发明涉及一种金属元素晶格掺杂于si基材料的催化剂及其在无氧条件下催化甲烷直接制备乙烯的方法,该过程实现甲烷的零积碳转化和乙烯的高选择性合成。
背景技术:
天然气(甲烷)资源的开发和有效利用代表着当代能源结构的发展方向,也是可持续发展的保证和能源绿色化的重要途径之一。近年来西方发达发国家在页岩气以及“可燃冰”的开发方面亦取得了突破性的进展,爆发了一场“页岩气革命”。我国页岩气资源类型多、分布相对集中,可采资源潜力为25万亿立方米(不含青藏区),与我国陆域常规天然气相当,与美国的24万亿立方米相近,国家“十二五”期间在页岩气开发领域已进行的部署,欲在几个不同类型的页岩油气区取得技术上的突破并初步建立有经济效益的生产能力。
然而,如何高效利用气态碳氢资源(甲烷)已成为制约我国能源工业发展的重要环节,将这种丰富的资源转化为燃料和高附加值的化学品(特别是低碳烯烃)重新激起了世界范围的兴趣,同时也是改善我国能源结构的重要步骤。低碳烯烃,诸如乙烯等是化学化工过程中非常重要的原料或中间体,传统低碳烯烃(c2-c4)主要来源于石脑油裂解等石化过程,从而乙烯的生产已成为衡量一个国家和地区石油化工生产水平的标志。随着石油资源的日益枯竭,探索非传统路线制低碳烯烃的方法已经成为当前研究关注的焦点。随之一些典型的替代路线应运而生,如从合成气出发经甲醇或二甲醚,进一步转化获得低碳烯烃,但该路线过程复杂,原子经济性较低。为了缩短反应路径,从合成气出发经费托(fischer–tropsch)路线直接合成低碳烯烃也进行了大量的研究。但是上述的替代路线都必须消耗co或h2来除去co中的o,将不可避免地造成c的原子利用率低于50%。尽管高昂的产能投入、大量的co2排放以及低于50%原子利用率,但间接过程仍在天然气工业应用中占据主导地位。
与此相反,天然气的直接转化则具有巨大的经济潜力且更为环保。然而对于天然气的直接转化仍然是化学、化工过程中的难题。天然气的主要成分为甲烷,其c—h键键能高达434kj/mol,而甲烷分子本身几乎不存在电子亲和性,此外其电离能大而极化率小,所以甲烷的c—h键活化被认为是化学乃至化学领域的“圣杯”。keller和bhasin报导了在o2参与下实现了甲烷c—h键的活化,二位富有开创性的工作燃起了世界范围内研究高温(>1073k)条 件下甲烷氧化偶联制乙烯的热情,其间人们合成并测试了数百种催化材料,在上世纪90年代研究达到顶峰。氧化偶联过程中由于分子氧(o2)的引入,不可避免地导致甲烷及其产物的过度氧化,从而产生大量热力学比甲烷更稳定的产物,如co2和h2o,最终导致c原子的利用效率相对较低。由于新材料和新催化剂开发的瓶颈致使甲烷氧化偶联过程发展停滞不前,到目前为止具有经济可行性的新工艺仍鲜为报道。最近的一项研究提出利用弱氧化性的气相s替代分子氧o2,进行甲烷氧化偶联反应。在温度为1323k时(反应气:5%ch4/ar),最优的pds/zro2催化剂可以实现16%的甲烷转化率,然而c2h4的选择性仅为20%左右,却副产大量cs2和h2s。以上研究表明,氧(或氧化剂)助甲烷活化均将不可避免的导致过氧化发生。
因此,无氧(或无氧化剂)直接转化甲烷被视为是最为理想的甲烷活化转化路线。无氧(或无氧化剂)条件下,能够有效地避免甲烷或产物的过度氧化,抑制了温室气体co2的排放,进而提高了c原子的利用率。甲烷直接催化转化制乙烯的挑战在于:1)可控活化甲烷使其第一个c—h键断裂;2)抑制其在催化剂表面的深度脱氢;3)避免产生温室气体co2和积碳。其中1、2针对催化剂,而3针对反应过程。对于有氧过程产物的过氧化是不可避免的,致使co2的产生也是不可避免的。唯有无氧过程才能避免co2的产生,但其易于积碳,因此研究如何避免积碳就成为无氧过程目前关注的焦点。解决积碳问题关键在了解积碳的来源,以无氧芳构化过程为例,其积碳主要来源于:甲烷在催化剂的mo物种表面深度脱氢积碳(“类石墨化积碳”);产物扩散过程中在载体分子筛孔道或孔口的b酸性位上环化偶联积碳(“聚芳烃积碳”)。因此甲烷直接转化制乙烯的三个挑战均在于催化剂的设计与构建。
1993年,大连化物所的研究人员首次报道了mo/hzsm-5催化剂上连续流动模式下ch4无氧芳构化反应。在973k和常压下,ch4转化率大约为6%,芳烃的选择性大于90%(不计反应积炭),成为ch4无氧芳构化过程研究的重要里程碑。在过去的十几年里,多国科学家的研究工作主要集中在催化剂的制备与开发、反应和失活机理等方面,但催化剂快速积碳失活制约其进一步工业放大。
最近,美国siluria公司(us201241246、us2013165728、us2014121433、ca2837201、us8921256b29),利用生物模版法制备的复合催化剂在600-650℃氧化偶联反应中甲烷转化率为26%,乙烯选择性为52%,目前该公司正在进行中试,预计2017-2018将进行工业化示范。对于甲烷选择氧化制备甲醇或甲醛,由于目标产物甲醇和甲醛的氧化速度要比原料甲烷快得多,导致反应的选择性较低,基本不具有规模应用前景。
对于申请号为201310174960.5专利所公开的方法中,所述的催化剂,金属元素在si基材料中为晶格掺杂与表面负载共存,致使催化剂结构存在一些缺陷,从而导致催化剂和反应 过程呈现如下缺点:(1)氧化还原耐受温度仅为800~1150℃;(2)催化剂寿命较短(100小时左右);(3)乙烯选择性偏低(30-90%);(4)芳烃产物选择性偏高(10-70%);(5)反应初期有少量积碳。
技术实现要素:
本发明技术解决问题:针对现有技术中存在问题,经过长期对比研究,本发明改进了催化剂制备过程,提供一种金属元素晶格掺杂si基材料催化剂的制备方法及甲烷无氧制乙烯的方法,实现了金属元素全部晶格掺杂于si基材料,极大的提高了氧化还原耐受温度、催化剂寿命、乙烯选择性,同时降低了芳烃选择性,实现整个过程零积碳。
本发明技术解决方案:
本发明涉及一种制备晶格掺杂的催化剂及其在无氧条件下催化甲烷直接制备乙烯的方法。所谓的甲烷无氧转化是指在无分子氧(o2)或无单质硫(s)或无氧硫化合物(如,so2等)存在的条件下直接将甲烷转化的方式。
所述的催化剂为单原子金属晶格掺杂于si基化合物晶格中的熔融体无定形催化剂,所述的掺杂为晶格掺杂;所谓的晶格掺杂是指掺杂金属元素与基质材料中某些元素形成化学键,使掺杂金属元素被限制于掺杂基质的晶格中,从而产生特定的催化性能。
按催化剂的总重量为100%计,金属元素晶格掺杂的催化剂中金属掺杂量应大于10ppm,小于等于1.0wt.%。
该催化剂将甲烷为原料气直接催化反应转化为乙烯,同时联产丙烯、丁烯、芳烃和氢气。
所谓的熔融体无定形材料,在催化剂制备过程中金属和硅基材料为全部熔融态,极速冷却后而形成具有长程无序和短程也无序的无定形材料。
所述的掺杂金属元素包括:锂、镁、钙、锶、钡、锰、镧、铬、钼、钨、铼、铁、钴、镍、锌、镓、金、铂、钌、钯、银,优选,锂、镁、钙、锶、镧、钡、锰、钴、铁、镍、锌、镓、金、银、钯。
所述的掺杂金属元素,掺杂金属的存在状态为氧化物、碳化物、硅化物的一种或多种;
所述催化剂是以si与c、o中的一种或两种以上的硅基材料为主体,于其中晶格掺杂有金属元素形成熔融态固化后获得的。
所述掺杂金属元素的前驱体(预掺杂金属元素的存在状态)包括:硝酸盐、硫酸盐、碳酸盐、氯化物、c数为1~2的有机酸盐、c数为1~2的有机醇盐中的一种或二种以上;
所述掺杂金属元素的硅基材料,其硅源包括液体硅源、固体硅源和气体硅源;
所述液体硅源优选但不限于,硅酸四乙酯、四氯化硅、四溴化硅、三氯氢硅。
所述固体硅源优选但不限于,二氧化硅、碳化硅、氮化硅、碘化硅、单质硅中的一种或二种以上;固体硅源的粒径优选10nm-2μm;硅源比表面积优选1-300m2/g。
所述气体硅源优选但不限于,硅烷(四氢化硅)。
所述催化剂的采用下述固相掺杂技术中的任一一种制备获得:
以下制备过程的目的是提高金属元素在硅基材料中的分散度,同时更有效的使金属元素晶格掺杂于si与c、o中的一种或两种以上形成的熔融体无定形材料中。
所述固相掺杂技术包括:改进化学气相沉积法(mcvd)、溶胶-凝胶配合改进的化学气相沉积法(mcvd)、多孔质si化合物浸润配合改进的化学气相沉积法(mcvd)等固相掺杂技术;
改进的化学气相沉积法(mcvd):在特定温度40~220℃下,将四氯化硅液体和气相掺杂金属或挥发性掺杂金属盐(金属氯化物、c数为1~2的有机醇盐和c数为1~2的有机酸盐中的一种或二种以上)在载气(99.999%的高纯氧)带动下,进入mcvd装置在1400-1650℃进行反应,并气相沉积,随后在1800~2200℃熔制获得相应金属掺杂的硅基催化剂,然后立即冷却(气体冷却、水冷却、液氮冷却),固化后得催化剂;
溶胶-凝胶配合改进的化学气相沉积法(mcvd):将四氯化硅液体在载气(高纯氧)带动下,进入mcvd装置在1400-1650℃进行反应,并气相沉积,将获得的沉积材料液相浸渍于正硅酸四乙酯(teos)和待掺杂金属盐(可溶性金属氯化物、硝酸盐、硫酸盐、碳酸盐)水溶液中进行水解反应,随后在1800~2200℃熔制获得相应金属掺杂的硅基催化剂,然后立即冷却(气体冷却、水冷却、液氮冷却、油冷却),固化后得催化剂。
多孔质si化合物浸润配合改进的化学气相沉积法(mcvd):将四氯化硅液体在载气(99.999%的高纯氧)带动下,进入mcvd装置在1400-1650℃进行反应,并气相沉积,将获得的多孔质si化合物液相浸渍于待掺杂金属盐(可溶性金属氯化物、硝酸盐、硫酸盐、碳酸盐)水溶液中,随后在1800~2200℃熔制获得相应金属掺杂的硅基催化剂,然后立即冷却(气体冷却、水冷却、液氮冷却,油冷却),固化后得催化剂。
溶胶-凝胶配合改进的化学气相沉积法(mcvd)中包括两个重要的步骤即溶胶-凝胶过程和浸渍过程,其中溶胶-凝胶时间优选6-24小时;溶胶-凝胶温度为50-90℃;溶胶-凝胶ph值优选1-5。随后的浸渍过程,浸渍时间优选1-6小时;浸渍温度为室温;浸渍的ph值优选8-10。
多孔质si化合物浸润配合改进的化学气相沉积法(mcvd)中包括一个重要的步骤,浸渍时间优选2-16小时;温度为20-50℃,优选常温;ph值优选5-8。
所述固化是指催化剂制备过程中熔制后物料的一个重要的冷却过程,所述的冷却为快速 冷却或自然冷却;
快速冷却方式包括气体冷却、水冷却、液氮冷、油冷却中的一种或二种以上组合;快冷却速率优选50℃/s~100,000,000℃/s,优选;100~10,000,000℃/s,进一步优选:500-1,000,000℃/s。
油冷却采用的油的种类包括;矿物油(饱和烃含量50~95%、s含量≤0.03%、粘度指数(vi)为80~170)、菜籽油、硅油、pao(聚α烯烃)中的一种或二种以上;气体冷却中的气体为惰性气体(氦气、氩气)、氮气、氧气或空气中的一种或两种以上。
熔制后固化获得的熔融态无定形催化剂需进行粉碎或造型的过程;
粉碎后催化剂的粒径优选100nm~2cm;
所谓造型就是将熔制后获得熔融态无定形催化剂进行加工制作,来获得满足各种反应过程的规格式样(如,蜂窝煤状整体式催化剂),或直接制作成列管反应器进行反应(不需另添加催化剂)。
所述金属元素晶格掺杂于si与c、o中的一种或两种以上形成的熔融体无定形催化剂可以表示为
所述的
本发明涉及一种甲烷无氧直接制乙烯的方法,反应模式可以为流化床、移动床或固定床。
本发明涉及一种甲烷无氧直接制乙烯的方法,反应原料气体组成包括除甲烷之外,还可能包括惰性气氛气体和非惰性气氛气体中的一种或两种;惰性气氛气体为氮气、氦气、氩气中的一种或二种以上,惰性气氛气体在反应原料气中的体积含量为0~95%;非惰性气氛气体为一氧化碳、氢气、二氧化碳、水、一元醇(c数为2~4)、或c数为2~4烷烃中的一种或二种以上的混合物,非惰性气氛气体与甲烷的体积含量比为0~10%;反应原料气体中甲烷的体积含量为5~100%。
本发明涉及一种甲烷无氧直接制烯烃的方法,该反应过程中包括一个催化剂预处理过程,预处理气氛为反应原料气、氢气或空气;预处理温度为750~900℃;预处理压力0.1~1mpa;反应原料气的质量空速为0.8~2.5l/g/h,优选1.0~2.0l/g/h。
本发明涉及一种甲烷无氧直接制乙烯的方法,该反应过程为连续流动反应模式或间歇反 应模式。连续流动反应模式:反应温度为750~1150℃,优选900~1100℃;反应压力为0.1~1mpa,优选0.1~0.5mpa;反应原料气的质量空速为1000~30000ml/g/h,优选4000~20000ml/g/h;间歇反应模式:反应压力优选1~20mpa;反应时间≥5分钟。
本发明涉及一种甲烷无氧直接制乙烯的方法,该过程还联产丙烯、丁烯、芳烃和氢气,芳烃产物包括苯、甲苯、对二甲苯、邻二甲苯、间二甲苯、乙苯、萘中的一种或多种。
本发明基于长期甲烷无氧芳构化研究的基础上,提出利用金属晶格掺杂的硅基催化剂在无氧条件下催化甲烷直接制取乙烯、芳烃和氢气的方法,该方法与先前的甲烷无氧转化过程,尤其与申请号为201310174960.5专利相比具有如下特点:
虽然该过程的产物类型与现有的甲烷无氧芳构化过程比较接近,但研究发现二者存在本质的区别(催化剂和反应机理)。首先,甲烷无氧芳构化的催化剂为分子筛负载型催化剂;其次,目前公认的甲烷无氧芳构化的反应机理(如式1所示):甲烷在催化剂的活性物种(mocx、wc、re)表面发生解离生成chx物种,随后chx物种在催化剂表面进行偶联生成c2hy物种,该物种进一步在分子筛孔道的酸性位上进行偶联,同时经过分子筛孔道的折形选择生成芳烃(j.energychem.2013,22,1-20)。
式1mocx/zeolite催化剂催化甲烷无氧芳构化的反应机理。
然而本发明的催化剂为金属元素晶格掺杂于si与c、o中的一种或两种以上形成的熔融态无定形材料;反应机理为甲烷经过活性物种(晶格中的化合态金属元素)的诱导生成甲基自由基(·ch3),随后甲基自由基进一步偶联脱氢获得烯烃,并联产芳烃和氢气(如式2所示)。
式2
无氧芳构化过程与本发明过程存在以下差异:1)具有特定孔道尺寸和结构、酸性位数量和种类的分子筛是芳构化过程所必须的;2)本发明的催化剂为熔融体无定形的无孔道、无酸性材料;3)芳构化机理为活性物种和分子筛(孔道和酸性)的协同催化机理,而本发明为自由基诱导机理。
本发明甲烷的转化率为10~70%;乙烯选择性为60~95%;丙烯和丁烯的选择性为5-25%;芳烃选择性为0~25%;零积碳。本发明具有催化剂寿命长(>500h)、催化剂高温(<1400℃)下氧化还原和水热稳定性好、乙烯选择性高、零积碳、产物易于分离、过程重复性好、操作安全可靠等特点,具有广阔的工业应用前景。
附图说明
图1为本发明制备方法的实现流程图;
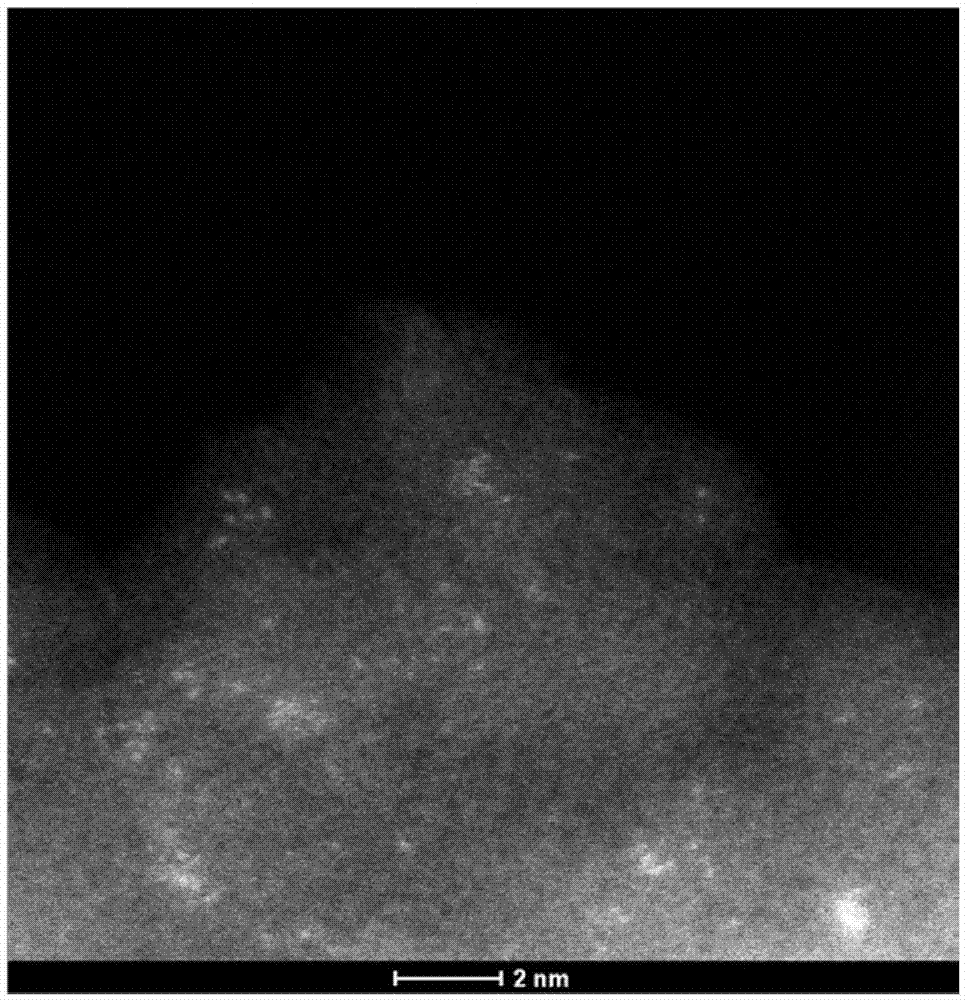
图2a-d为haadf-stem高分辨电镜照片和edx表征图,其中:图2a为0.02wt.%au-0.14wt.%
具体实施方式
下面结合附图及具体实施例详细介绍本发明。但以下的实施例仅限于解释本发明,本发明的保护范围应包括权利要求的全部内容,不仅仅限于本实施例。
如图1所示,本发明制备方法具体实现如下:
1.催化剂的制备
晶格掺杂催化剂制备方法包括化学气相沉积法(mcvd)、溶胶-凝胶掺杂法、多孔质si化合物浸润法等固相掺杂技术。催化剂标记为:
实施例1
改进化学气相沉积法(mcvd)
使用30ml/min的高纯氧将sicl4液体、fecl3水溶液鼓泡带入高温mcvd,在1600℃下sicl4和fecl3进行氧化沉积,获得fe掺杂的sio2粉体材料,随后在温度为2200℃、2bar纯氧气氛下熔制3h,然后迅速在冰水进行冷却,即获得0.2wt.%
实施例2
改进化学气相沉积法(mcvd)
使用30ml/min的高纯氧将sicl4液体、cocl2、nicl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、cocl2和nicl2进行氧化沉积,获得co、ni掺杂的sio2粉体材料,随后在温度为2200℃、2bar纯氮气氛下熔制3h,然后迅速在液氮中进行冷却,即获得0.5wt.%co-0.5wt.%
实施例3
改进化学气相沉积法(mcvd)
使用30ml/min的高纯氧将sicl4液体、pdcl2、licl水溶液鼓泡带入高温mcvd,在1600℃下sicl4、pdcl2和licl进行氧化沉积,获得pd、li掺杂的sio2粉体材料,随后在温度为2200℃、2bar纯氮气氛下熔制1.5h,然后迅速在液氮中进行冷却,即获得0.5wt.%pd-0.5wt.%
实施例4
改进化学气相沉积法(mcvd)
使用30ml/min的高纯氧将sicl4液体、aucl3、mgcl2水溶液鼓泡带入高温mcvd,在 1600℃下sicl4、aucl3和mgcl2进行氧化沉积,获得au、mg掺杂的sio2粉体材料,随后在温度为2200℃、2bar纯氮气氛下熔制1h,然后迅速在液氮中进行冷却,即获得0.2wt.%au-0.4wt.%
实施例5
改进化学气相沉积法(mcvd)
使用30ml/min的高纯氧将sicl4液体、bacl2、srcl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、bacl2和srcl2进行氧化沉积,获得ba、sr掺杂的sio2粉体材料,随后在温度为2200℃、2bar纯氮气氛下熔制3h,然后迅速在液氮中进行冷却,即获得0.1wt.%ba-0.3wt.%
实施例6
改进化学气相沉积法(mcvd)
使用30ml/min的高纯氧将sicl4液体、zncl2、cacl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、zncl2和cacl2进行氧化沉积,获得zn、ca掺杂的sio2粉体材料,随后在温度为2200℃、2bar纯氮气氛下熔制2h,然后迅速在液氮中进行冷却,即获得0.5wt.%zn-0.1wt.%
实施例7
溶胶-凝胶配合改进的化学气相沉积法(mcvd):
使用30ml/min的高纯氧将sicl4液体、fecl3水溶液鼓泡带入高温mcvd,在1600℃下sicl4和fecl3进行氧化沉积,获得fe掺杂的sio2粉体材料,随后在60℃下将上述粉体材料浸渍于正硅酸四乙酯(teos)和fe(no3)3的水溶液中进行水解反应约6小时;进而在温度为2200℃、2bar纯氧气氛下熔制3h,然后迅速在冰水进行冷却,即获得0.2wt.%
实施例8
溶胶-凝胶配合改进的化学气相沉积法(mcvd):
使用30ml/min的高纯氧将sicl4液体、cocl2、nicl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、cocl2和nicl2进行氧化沉积,获得co、ni掺杂的sio2粉体材料,随后在60℃下将上述粉体材料浸渍于正硅酸四乙酯(teos)、co(no3)2和ni(no3)2的水溶液中进行水解反应约8小时;进而在温度为2200℃、2bar纯氧气氛下熔制2h,然后迅速在冰水进行冷却,即获得0.5wt.%co-0.5wt.%
实施例9
溶胶-凝胶配合改进的化学气相沉积法(mcvd):
使用30ml/min的高纯氧将sicl4液体、fecl3、aucl3水溶液鼓泡带入高温mcvd,在1600℃下sicl4、fecl3和aucl3进行氧化沉积,获得fe、au掺杂的sio2粉体材料,随后在80℃下将上述粉体材料浸渍于正硅酸四乙酯(teos)、fe(no3)3和haucl4的水溶液中进行水解反应约8小时;进而在温度为2200℃、2bar氩气氛下熔制2h,然后迅速在冰水进行冷却,即获得0.3wt.%fe-0.1wt.%
实施例10
溶胶-凝胶配合改进的化学气相沉积法(mcvd):
使用25ml/min的高纯氧将sicl4液体、pdcl2、licl水溶液鼓泡带入高温mcvd,在1600℃下sicl4、pdcl2和licl进行氧化沉积,获得pd、li掺杂的sio2粉体材料,随后在80℃下将上述粉体材料浸渍于正硅酸四乙酯(teos)、pdcl2和li2so4的水溶液中进行水解反应约6小时;进而在温度为2200℃、2bar氩气氛下熔制2h,然后迅速在冰水进行冷却,即获得0.4wt.%pd-0.3wt.%
实施例11
溶胶-凝胶配合改进的化学气相沉积法(mcvd):
使用25ml/min的高纯氧将sicl4液体、aucl3、mgcl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、aucl3和mgcl2进行氧化沉积,获得au、mg掺杂的sio2粉体材料,随后在80℃下将上述粉体材料浸渍于正硅酸四乙酯(teos)、aucl2和mg(no3)2的水溶液中进行水解反应约12小时;进而在温度为2200℃、2bar氩气氛下熔制3h,然后迅速在氩气气氛下进行冷却,即获得0.2wt.%au-0.4wt.%
实施例12
溶胶-凝胶配合改进的化学气相沉积法(mcvd):
使用25ml/min的高纯氧将sicl4液体、bacl2、srcl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、bacl2和srcl2进行氧化沉积,获得ba、sr掺杂的sio2粉体材料,随后在70℃下将上述粉体材料浸渍于正硅酸四乙酯(teos)、srcl2和ba(no3)2的水溶液中进行水解反应约12小时;进而在温度为2200℃、2bar氩气氛下熔制2h,然后迅速在氩气气氛下进行冷却,即获得0.1wt.%ba-0.3wt.%
实施例13
溶胶-凝胶配合改进的化学气相沉积法(mcvd):
使用30ml/min的高纯氧将sicl4液体、zncl2、cacl2水溶液液鼓泡带入高温mcvd,在1600℃下sicl4、zncl2和cacl2进行氧化沉积,获得zn、ca掺杂的sio2粉体材料,随后在80℃下将上述粉体材料浸渍于正硅酸四乙酯(teos)、zncl2和ca(no3)2的水溶液中 进行水解反应约12小时;随后在温度为2200℃、2bar纯氮气氛下熔制2h,然后迅速在液氮中进行冷却,即获得0.4wt.%zn-0.1wt.%
实施例14
多孔质si化合物浸润配合改进的化学气相沉积法(mcvd):
使用30ml/min的高纯氧将sicl4液体、fecl3水溶液鼓泡带入高温mcvd,在1600℃下sicl4和fecl3进行氧化沉积,获得fe掺杂的sio2粉体材料,随后在常温将上述粉体材料浸渍于fe(no3)3的水溶液中进行约12小时;进而在温度为2200℃、2bar纯氧气氛下熔制3h,然后迅速在冰水进行冷却,即获得0.2wt.%
实施例15
多孔质si化合物浸润配合改进的化学气相沉积法(mcvd):
使用30ml/min的高纯氧将sicl4液体、cocl2、nicl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、cocl2和nicl2进行氧化沉积,获得co、ni掺杂的sio2粉体材料,随后在常温下将上述粉体材料浸渍于co(no3)2和ni(no3)2的水溶液中进行约8小时;进而在温度为2200℃、2bar纯氧气氛下熔制2h,然后迅速在冰水进行冷却,即获得0.05wt.%co-0.2wt.%
实施例16
多孔质si化合物浸润配合改进的化学气相沉积法(mcvd):
使用30ml/min的高纯氧将sicl4液体、fecl3、aucl3水溶液鼓泡带入高温mcvd,在1600℃下sicl4、fecl3和aucl3进行氧化沉积,获得fe、au掺杂的sio2粉体材料,随后在常温将上述粉体材料浸渍于fe(no3)3和haucl4的水溶液中约8小时;进而在温度为2200℃、2bar氩气氛下熔制2h,然后迅速在冰水进行冷却,即获得0.3wt.%fe-0.1wt.%
实施例17
多孔质si化合物浸润配合改进的化学气相沉积法(mcvd):
使用25ml/min的高纯氧将sicl4液体、pdcl2、licl水溶液鼓泡带入高温mcvd,在1600℃下sicl4、pdcl2和licl进行氧化沉积,获得pd、li掺杂的sio2粉体材料,随后在常温将上述粉体材料浸渍于pdcl2和li2so4的水溶液中约6小时;进而在温度为2200℃、2bar氩气氛下熔制2h,然后迅速在冰水进行冷却,即获得0.04wt.%pd-0.03wt.%
实施例18
多孔质si化合物浸润配合改进的化学气相沉积法(mcvd):
使用25ml/min的高纯氧将sicl4液体、aucl3、mgcl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、aucl3和mgcl2进行氧化沉积,获得au、mg掺杂的sio2粉体材料,随后在常温将上述粉体材料浸渍于aucl3和mg(no3)2的水溶液中约12小时;进而在温度为2200℃、2bar氩气氛下熔制3h,然后迅速在冰水下进行冷却,即获得0.02wt.%au-0.14wt.%
实施例19
多孔质si化合物浸润配合改进的化学气相沉积法(mcvd):
使用25ml/min的高纯氧将sicl4液体、bacl2、srcl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、bacl2和srcl2进行氧化沉积,获得ba、sr掺杂的sio2粉体材料,随后在50℃下将上述粉体材料浸渍于srcl2和ba(no3)2的水溶液中进行水解反应约2小时;进而在温度为2200℃、2bar氩气氛下熔制2h,然后迅速在液氮下进行冷却,即获得0.01wt.%ba-0.3wt.%
实施例20
多孔质si化合物浸润配合改进的化学气相沉积法(mcvd):
使用30ml/min的高纯氧将sicl4液体、zncl2、cacl2水溶液液鼓泡带入高温mcvd,在1600℃下sicl4、zncl2和cacl2进行氧化沉积,获得zn、ca掺杂的sio2粉体材料,随后在常温将上述粉体材料浸渍于zncl2和ca(no3)2的水溶液中进行水解反应约2小时;随后在温度为2200℃、2bar纯氮气氛下熔制2h,然后迅速在液氮中进行冷却,即获得0.4wt.%zn-0.01wt.%
实施例21
改进化学气相沉积法(mcvd)
使用30ml/min的高纯氧将sicl4液体、fecl3水溶液鼓泡带入高温mcvd,在1600℃下sicl4和fecl3进行氧化沉积,获得fe掺杂的sio2粉体材料,随后在温度为2200℃、2bar纯氧气氛下熔制3h,然后在1500℃下通入ch4进行碳化2h,迅速在冰水进行冷却,即获得0.2wt.%
实施例22
改进化学气相沉积法(mcvd)
使用30ml/min的高纯氧将sicl4液体、cocl2、nicl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、cocl2和nicl2进行氧化沉积,获得co、ni掺杂的sio2粉体材料,随后在温度为2200℃、2bar纯氮气氛下熔制3h,然后在1500℃下通入ch4进行碳化2h,迅速在液氮中进行冷却,即获得0.5wt.%co-0.5wt.%
实施例23
溶胶-凝胶配合改进的化学气相沉积法(mcvd):
使用25ml/min的高纯氧将sicl4液体、bacl2、srcl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、bacl2和srcl2进行氧化沉积,获得ba、sr掺杂的sio2粉体材料,随后在70℃下将上述粉体材料浸渍于正硅酸四乙酯(teos)、srcl2和ba(no3)2的水溶液中进行水解反应约12小时;进而在温度为2200℃、2bar氩气氛下熔制2h,然后在1500℃下通入ch4进行碳化2h,迅速在液氮下进行冷却,即获得0.1wt.%ba-0.3wt.%
实施例24
溶胶-凝胶配合改进的化学气相沉积法(mcvd):
使用30ml/min的高纯氧将sicl4液体、zncl2、cacl2水溶液液鼓泡带入高温mcvd,在1600℃下sicl4、zncl2和cacl2进行氧化沉积,获得zn、ca掺杂的sio2粉体材料,随后在80℃下将上述粉体材料浸渍于正硅酸四乙酯(teos)、zncl2和ca(no3)2的水溶液中进行水解反应约12小时;随后在温度为2200℃、2bar纯氮气氛下熔制2h,然后在1500℃下通入ch4进行碳化3h,迅速在液氮中进行冷却,即获得0.4wt.%zn-0.1wt.%
实施例25
多孔质si化合物浸润配合改进的化学气相沉积法(mcvd):
使用25ml/min的高纯氧将sicl4液体、aucl3、mgcl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、aucl3和mgcl2进行氧化沉积,获得au、mg掺杂的sio2粉体材料,随后在常温将上述粉体材料浸渍于aucl3和mg(no3)2的水溶液中约12小时;进而在温度为2200℃、2bar氩气氛下熔制3h,然后在1500℃下通入ch4进行碳化3h,迅速在液氮下进行冷却,即获得0.02wt.%au-0.14wt.%
实施例26
多孔质si化合物浸润配合改进的化学气相沉积法(mcvd):
使用25ml/min的高纯氧将sicl4液体、bacl2、srcl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、bacl2和srcl2进行氧化沉积,获得ba、sr掺杂的sio2粉体材料,随后在50℃下将上述粉体材料浸渍于srcl2和ba(no3)2的水溶液中进行水解反应约1小时;进而在温度为2200℃、2bar氩气氛下熔制2h,然后在1500℃下通入c2h4进行碳化3h,迅速在冰水下进行冷却,即获得0.01wt.%ba-0.3wt.%
实施例27
改进化学气相沉积法(mcvd):
使用30ml/min的高纯氧将sicl4液体、cocl2、nicl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、cocl2和nicl2进行氧化沉积,获得co、ni掺杂的sio2粉体材料,随后在温度为2200℃、2bar纯氮气氛下熔制3h,然后在1500℃下通入ch4进行碳化1h,迅速在液氮中进行冷却,即获得0.3wt.%co-0.5wt.%
实施例28
溶胶-凝胶配合改进的化学气相沉积法(mcvd):
使用25ml/min的高纯氧将sicl4液体、bacl2、srcl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、bacl2和srcl2进行氧化沉积,获得ba、sr掺杂的sio2粉体材料,随后在70℃下将上述粉体材料浸渍于正硅酸四乙酯(teos)、srcl2和ba(no3)2的水溶液中进行水解反应约12小时;进而在温度为2200℃、2bar氩气氛下熔制2h,然后在1500℃下通入ch4进行碳化1h,迅速在液氮下进行冷却,即获得0.1wt.%ba-0.3wt.%
实施例29
溶胶-凝胶配合改进的化学气相沉积法(mcvd):
使用30ml/min的高纯氧将sicl4液体、zncl2、cacl2水溶液液鼓泡带入高温mcvd,在1600℃下sicl4、zncl2和cacl2进行氧化沉积,获得zn、ca掺杂的sio2粉体材料,随后在80℃下将上述粉体材料浸渍于正硅酸四乙酯(teos)、zncl2和ca(no3)2的水溶液中进行水解反应约12小时;随后在温度为2200℃、2bar纯氮气氛下熔制2h,然后在1500℃下通入ch4进行碳化1h,迅速在液氮中进行冷却,即获得0.4wt.%zn-0.1wt.%
实施例30
多孔质si化合物浸润配合改进的化学气相沉积法(mcvd):
使用25ml/min的高纯氧将sicl4液体、aucl3、mgcl2水溶液鼓泡带入高温mcvd,在1600℃下sicl4、aucl3和mgcl2进行氧化沉积,获得au、mg掺杂的sio2粉体材料,随后在常温将上述粉体材料浸渍于aucl3和mg(no3)2的水溶液中约12小时;进而在温度为2200℃、2bar氩气氛下熔制3h,然后在1500℃下通入ch4进行碳化0.5h,迅速在液氮下进行冷却,即获得0.02wt.%au-0.14wt.%
催化剂的制备不限于实施例所述的金属元素。
2.催化剂表征
1)电感耦合等离子体发射光谱(icp-aes)表征
利用电感耦合等离子体发射光谱仪(icp-aes)酸洗(硝酸和hf酸)方法,所谓的酸洗 icp过程:如果金属负载于载体表面,我们可以使用酸洗过程将载体表面的金属溶解(该酸只能溶解金属而不能溶解载体),通过icp测量可以获得酸洗的程度(即表面负载量/(表面负载量+掺杂量)),而如果金属元素不能被酸溶解,说明金属元素已掺入硅基基质晶格而被保护。首先我们使用稀硝酸酸洗0.5wt.%
2)负载型0.5wt.%zn-0.3%sr/sio2(催化剂制备实施例31)催化剂的酸洗icp表征
首先使用稀硝酸酸洗0.5wt.%co/sio2催化剂,icp分析结果表明全部zn、sr离子被溶解,而且量正好折算为其担载量,该结果表明zn和sr全部分散于硅基基质的表面而没有进入基质晶格。
3)0.5wt.%
图2a中,0.02wt.%au-0.14wt.%
3.在无氧连续流动条件下直接转化甲烷为烯烃、芳烃和氢气
以上所述所有催化剂在使用之前,均需粉碎过筛至20-30目备用。
所有反应实例均在连续流动微反应装置中进行,该装置配备气体质量流量计、气体脱氧脱水管和在线产物分析色谱(反应器的尾气直接与色谱的定量阀连接,进行周期实时采样分析)。除特殊说明之外,反应原料气均由10vol.%n2和90vol.%ch4组成,n2作为内标气。在线产物分析使用agilent7890b气相色谱配备fid和tcd双检测器,其中fid检测器配备hp-1毛细管柱对低碳烯烃、低碳烷烃和芳烃进行分析;tcd检测器配备hayesepd填充柱对低碳烯烃、低碳烷烃、甲烷、氢气和内标氮气进行分析。根据专利(cn1247103a,cn1532546a)计算甲烷转化率、含碳产物选择性和积炭,依据反应前后的碳平衡。
实施例1
将0.5g催化剂0.2wt.%
实施例2~12
将0.5g催化剂0.2wt.%au-0.4wt.%
实施例13-23
将1.5g催化剂0.3wt.%fe-0.1wt.%
实施例24~34
将0.75g催化剂0.4wt.%zn-0.01wt.%
实施例35
将0.6g催化剂0.01wt.%ba-0.3wt.%
实施例36
将0.65g催化剂0.2wt.%
实施例37-47
将0.75g催化剂0.3wt.%co-0.5wt.%
实施例48
将0.55g催化剂0.4wt.%zn-0.01wt.%
实施例49
将0.65g催化剂0.4wt.%zn-0.01wt.%
实施例50
将0.65g催化剂0.4wt.%zn-0.01wt.%
综上,本发明在固定床反应模式下,反应温度为750~1100℃;反应压力为常压;甲烷的质量空速为1.0~30.0l/g/h。甲烷的转化率为10~70%;烯烃选择性为60~95%;丙烯、丁烯的选择性为5~25%;芳烃选择性为0~25%。
由此得出结论:具有催化剂寿命长(>500h)、催化剂高温(<1400℃)下氧化还原和水热稳定性好、产物选择性高、零积碳、产物易于分离、过程重复性好、操作安全可靠等特点,具有广阔的工业应用前景。
需要说明的是,按照本发明上述各实施例,本领域技术人员是完全可以实现本发明独立权利要求及从属权利的全部范围的,实现过程及方法同上述各实施例;且本发明未详细阐述部分属于本领域公知技术。
以上所述,仅为本发明部分具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本领域的人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!