废气净化催化剂及其制造方法与流程
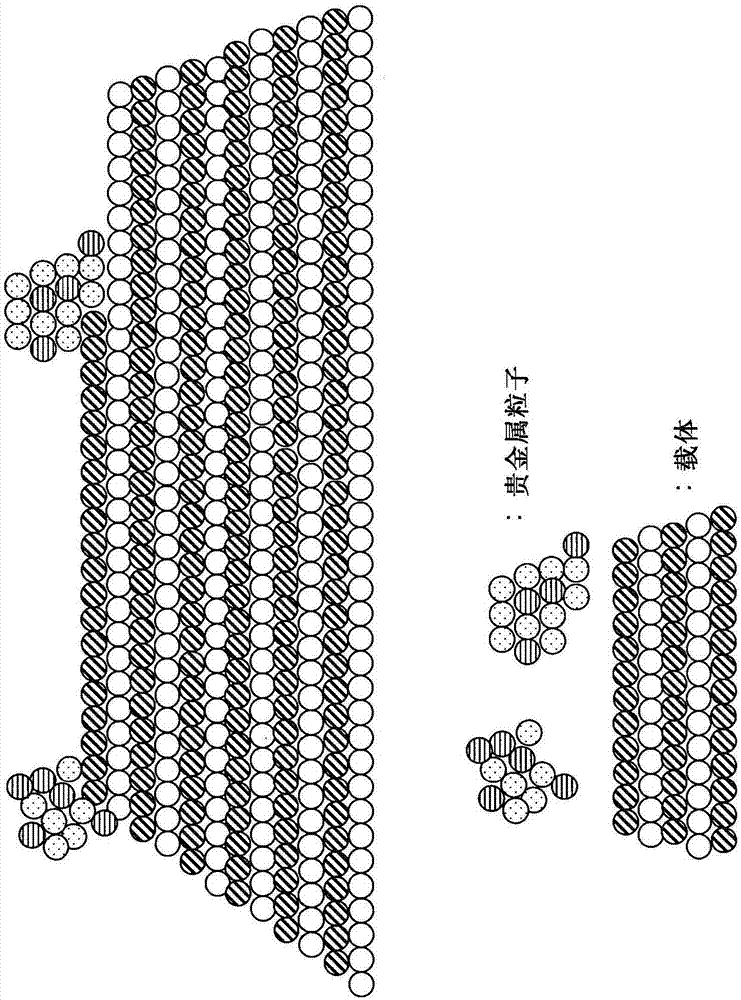
本申请是申请号为201080030713.2(国际申请号为pct/jp2010/062024)、中国国家阶段进入日为2012年1月9日(国际申请日为2010年7月9日)、发明名称为“废气净化催化剂及其制造方法”的中国发明专利申请的分案申请。本发明涉及废气净化催化剂及其制造方法。本发明特别涉及用于净化来自汽车等的内燃机的废气的废气净化催化剂及其制造方法。
背景技术:
:在来自汽车发动机等内燃机的废气中含有氮氧化物(nox)、一氧化碳(co)、碳氢化合物(hc)等。这些物质可利用对co及hc进行氧化且对nox进行还原的废气净化催化剂来净化。作为废气净化催化剂的代表,已知有使如铂、铑及钯这样的贵金属负载于如氧化铝这样的多孔金属氧化物载体上而得到的催化剂。对于使用这种贵金属的废气净化催化剂而言,在催化剂使用期间,贵金属粒子在载体上移动,多个贵金属粒子结合从而发生粒子生长的现象,即称为贵金属的烧结(结块)的现象成为问题。关于该问题,在专利文献1中提出了如下方案,即,使构成载体的钙钛矿型复合氧化物层自贵金属层外延生长,由此将由钙钛矿型复合氧化物构成的载体和贵金属强固地结合,从而抑制贵金属的烧结。具体而言,在该文献中,通过在基底基材上形成结晶性的贵金属层,并且在该贵金属层的表面形成钙钛矿型复合氧化物的外延载体层,然后将这些贵金属层及载体层的层叠体从基底基材分离,由此制造废气净化催化剂。此外,在该文献中,将这样得到的贵金属层及载体层的层叠体粉碎,得到由10nm~20nm大小的载体层和负载于该载体层上的1nm~2nm大小的催化剂层构成的粉末状的废气净化催化剂。对于该方法而言,由于将贵金属层及载体层的层叠体粉碎而得到粉末状的废气净化催化剂,因此在所得到的废气净化催化剂中,仅在载体的外表面负载有贵金属。另外,对于使用贵金属的废气净化催化剂而言,通常使贵金属高分散地负载于载体上来增大贵金属的表面积,这点对于废气的净化而言是优选的。但是,如图6(a)所示的废气净化催化剂10a那样,存在如下问题,即,在初期负载于载体11的各个贵金属粒子12a非常小、例如为原子水平的情况下,在催化剂的使用期间,贵金属粒子以无法控制的水平凝聚而发生粒子生长;贵金属以比较高的氧化状态存在于载体上,由此无法充分地得到催化活性。为了解决贵金属粒子过小造成的这些问题,已知有将控制了尺寸的贵金属粒子负载于载体上的技术(专利文献2~4)。关于贵金属粒子的尺寸控制,已知有如下技术,即,在溶液中形成控制了尺寸的贵金属胶体,其后,将该贵金属胶体负载于载体上。但是,如图6(b)所示,对于使用贵金属胶体而得到的现有的废气净化催化剂10b而言,存在如下问题,即,由贵金属胶体形成的贵金属粒子12b难以固定在载体11上,由此,在废气净化催化剂的使用期间,贵金属粒子12b会在载体11的表面移动,导致粒子生长。与此相关,在专利文献4中,如图6(c)所示,使由贵金属胶体形成的贵金属粒子12c部分地埋没于载体11中,由此,在废气净化催化剂10c的使用期间,抑制了贵金属粒子12c在载体11的表面移动而发生粒子生长的问题。但是,对于该方法而言,由于埋没于载体11中的部分贵金属不能与废气接触,不作为活性部位来发挥作用,因而造成浪费,因此,必须使用比较大量的贵金属。在此,还已知如下技术,即,在废气净化催化剂的使用期间,为了抑制贵金属粒子在载体表面移动而发生粒子生长,利用载体和负载于载体上的贵金属之间的化学亲和性。与此相关,例如,在专利文献5中提出了贵金属通过载体表面的氧与载体的阳离子键合形成表面氧化物层来抑制贵金属的粒子生长的技术,作为这种阳离子,特别列举出具有比锆小的电负性的阳离子。另外,在专利文献6中,提出了如下技术,即,在废气净化催化剂中,废气、贵金属微粒和载体粒子的三相界面对于废气的净化高效地发挥着功能,着眼于这一点,为了使该三相界面增多,利用真空电弧蒸镀装置,通过洛伦兹力及库仑力使贵金属离子与载体碰撞而附着并凝聚于载体,由此,在载体上生成近似半球状的贵金属粒子。如专利文献6那样,在利用真空电弧蒸镀装置使贵金属粒子堆积在载体上的情况下,难以进行贵金属粒径的控制,另外,虽然能够使贵金属粒子堆积于载体的外表面,但难以使贵金属粒子负载于载体的内部表面、即从载体的外侧不能直接观察的表面,例如,难以使贵金属粒子负载于细孔内的表面。另外,在利用真空电弧蒸镀装置的废气净化催化剂的制造方法中,难以以实用的速度制造废气净化催化剂。现有技术文献专利文献1:日本特开2008-279365号公报专利文献2:日本特开2006-314885号公报专利文献3:日本特开2008-55418号公报专利文献4:日本特开2007-812号公报专利文献5:日本特开2007-289920号公报专利文献6:日本特开2008-308735号公报技术实现要素:发明要解决的问题在本发明中,提供一种负载于载体上的贵金属粒子的粒子生长得到抑制的废气净化催化剂。另外,在本发明中,提供一种能够制造这种废气净化催化剂的废气净化催化剂的制造方法。进而,在本发明中,提供一种负载于载体上的贵金属粒子的粒子生长得到抑制,使用期间这种贵金属粒子的活性较高,且贵金属的使用量比较少的废气净化催化剂。并且在本发明中,提供一种能够制造这种废气净化催化剂的废气净化催化剂的制造方法。用于解决问题的方法本发明人发现,通过使贵金属粒子相对于结晶性金属氧化物载体外延生长且分散负载于载体的外表面和内表面、以及使特别控制了尺寸的贵金属粒子以近似半球状的形状负载于载体上,能够解决上述问题,从而想到了下述的本发明。〈1〉一种废气净化催化剂,具有结晶性金属氧化物载体及负载于所述载体上的贵金属粒子,所述贵金属粒子相对于所述载体外延生长,并且,所述贵金属粒子分散负载于所述载体的外表面和内表面。〈2〉如上述〈1〉所述的废气净化催化剂,其中,所述贵金属粒子以近似半球状的形状负载于所述载体上,所述载体与所述贵金属粒子相接的部分的宽度(w)和所述贵金属粒子距所述载体表面的高度(h)满足下式(1):w/h>1.0…(1),所述贵金属粒子距所述载体表面的高度(h)为0.5nm以上。〈3〉如上述〈1〉或〈2〉所述的废气净化催化剂,其中,所述贵金属粒子距所述载体表面的高度(h)为10.0nm以下。〈4〉如上述〈1〉~〈3〉中任一项所述的废气净化催化剂,其中,利用一氧化碳吸附法测定的所述贵金属粒子的粒径为0.5~10nm。〈5〉如上述〈1〉~〈4〉中任一项所述的废气净化催化剂,其中,所述贵金属选自由铂、钯、铑及它们的组合构成的组。〈6〉如上述〈1〉~〈5〉中任一项所述的废气净化催化剂,其中,所述载体选自由氧化铈、氧化锆、氧化铈-氧化锆固溶体、钙钛矿型金属氧化物、氧化钛及它们的组合构成的组。〈7〉如上述〈1〉~〈6〉中任一项所述的废气净化催化剂,其中,所述载体为金属氧化物载体,所述金属氧化物载体具有选自由碱金属、碱土金属、稀土类及它们的组合构成的组的金属元素。〈8〉如上述〈1〉~〈7〉中任一项所述的废气净化催化剂,其中,所述贵金属粒子距所述载体表面的高度(h)为5.0nm以下。〈9〉如上述〈1〉~〈8〉中任一项所述的废气净化催化剂,其中,在将下述的富气氛和贫气氛每2分钟切换一次、并在800℃下进行5小时的耐久以后,所述贵金属粒子的分布具有3.0nm以下的半峰全宽,富气氛:co-3%、h2o-5%、n2-剩余部分,贫气氛:o2-5%、h2o-5%、n2-剩余部分。〈10〉如上述〈1〉所述的废气净化催化剂,其中,所述贵金属粒子以近似半球状的形状负载于所述载体上,所述载体与所述贵金属粒子相接的部分的宽度(w)和所述贵金属粒子距所述载体表面的高度(h)满足下式(1):w/h>1.0…(1)所述贵金属粒子距所述载体表面的高度(h)为0.5nm以上且10nm以下,所述贵金属粒子为铂,并且所述载体为氧化铈或氧化铈-氧化锆固溶体。〈11〉如上述〈10〉所述的废气净化催化剂,其中,利用一氧化碳吸附法测定的所述贵金属粒子的粒径为1.0~5.0nm。〈12〉如上述〈1〉所述的废气净化催化剂,其中,所述贵金属粒子以近似半球状的形状负载于所述载体上,所述载体与所述贵金属粒子相接的部分的宽度(w)和所述贵金属粒子距所述载体表面的高度(h)满足下式(1):w/h>1.0…(1),所述贵金属粒子距所述载体表面的高度(h)为2nm以上且5nm以下,所述贵金属粒子为铑,并且所述载体为氧化铈或氧化铈-氧化锆固溶体。〈13〉如上述〈12〉所述的废气净化催化剂,其中,所述贵金属粒子和所述载体具有(111)面之间或(200)面之间的晶面接合。〈14〉一种废气净化装置,具有基材及保持在所述基材上的废气净化催化剂,并且,保持在所述基材上的所述废气净化催化剂中的、在所述基材的废气流上游侧负载的废气净化催化剂为上述〈1〉~〈13〉中任一项所述的废气净化催化剂。〈15〉如上述〈14〉所述的废气净化装置,其中,所述基材为蜂窝基材。〈16〉一种废气净化催化剂的制造方法,包括如下工序:在溶液中,由掩蔽剂掩蔽结晶性金属氧化物载体的至少一部分表面,将由掩蔽剂掩蔽了至少一部分表面的所述载体导入含有贵金属的含贵金属溶液中,然后,对所述载体及含贵金属溶液进行干燥及煅烧,使所述贵金属负载在所述载体上。〈17〉如上述〈16〉所述的方法,其中,所述掩蔽剂具有选自由仲氨基、叔氨基、羧基、羟基、羰基及它们的组合构成的组的官能团。〈18〉如上述〈16〉或〈17〉所述的方法,其中,所述含贵金属溶液为含有部分地还原和凝聚的贵金属凝聚体的含贵金属凝聚体溶液,并且,通过提供含有贵金属离子和/或配位化合物和聚合物的原料溶液而使所述贵金属离子和/或配位化合物配位于所述聚合物,然后使配位于所述聚合物的所述贵金属离子和/或配位化合物部分地还原和凝聚,从而得到所述含贵金属凝聚体溶液。〈19〉如上述〈16〉~〈18〉中任一项所述的方法,其中,通过加热、还原剂的添加或它们的组合来进行所述部分地还原和凝聚。〈20〉如上述〈16〉~〈19〉中任一项所述的方法,其中,所述聚合物具有选自由仲氨基、叔氨基、羧基、羟基、羰基及它们的组合构成的组的官能团。〈21〉如上述〈16〉~〈20〉中任一项所述的方法,其中,所述贵金属选自由铂、钯、铑及它们的组合构成的组。〈22〉如上述〈16〉~〈21〉中任一项所述的方法,其中,所述载体为金属氧化物载体,所述金属氧化物载体具有选自由碱金属、碱土金属、稀土类及它们的组合构成的组的金属元素。〈23〉一种废气净化催化剂,通过上述〈16〉~〈22〉中任一项所述的方法制造。附图说明图1是本发明的废气净化催化剂的概念剖面图。图2是现有技术的废气净化催化剂的概念剖面图。图3是使用掩蔽剂而得到的废气净化催化剂a-1(a)及不使用掩蔽剂而得到的废气净化催化剂a-2(b)的tem照片(实施例1)。图4是表示对废气净化催化剂a-1~a-3的碳氢化合物(hc)净化性能的评价结果的图(实施例1)。图5是控制贵金属粒子的形状及尺寸的废气净化催化剂的概念剖面图。图6是不控制贵金属粒子的形状及尺寸的废气净化催化剂的概念剖面图。图7是用于对控制贵金属粒子的形状及尺寸来制造废气净化催化剂的方法进行概念性地说明的图。图8是使用进行了1小时加热回流的含铂微粒溶液而得到的铂负载氧化铈催化剂(pt/ceo2)的tem照片(参考例1)。图9是使用进行了4小时加热回流的含铂微粒溶液而得到的铂负载氧化铈催化剂(pt/ceo2)的tem照片(参考例1)。图10是使用进行了8小时加热回流的含铂微粒溶液而得到的铂负载氧化铈催化剂(pt/ceo2)的tem照片(参考例1)。图11是使用进行了4小时加热回流的含铂微粒溶液而得到的铂负载氧化铝催化剂(pt/al2o3)的tem照片(参考例1)。图12是表示进行加热回流的时间和含铂粒子溶液的吸光度之间的关系的图(参考例2)。图13是实施例1和4以及参考例3和4所使用的试验装置的概略图。图14是表示耐久后的铂的平均粒径的图(参考例3)。图15是表示铂的平均粒径和碳氢化合物(hc)50%净化温度之间的关系的图(参考例4)。图16是表示铂的粒径和氧化状态之间的关系的图(参考例5)。图17是本发明的催化剂b-1的tem照片(实施例2)。图18是本发明的催化剂b-1的tem照片(实施例2)。图19是本发明的催化剂b-1的tem照片(实施例2)。图20是现有的催化剂b-2的tem照片(实施例2)。图21是现有的催化剂b-3的tem照片(实施例2)。图22是表示本发明的催化剂b-1及现有的催化剂b-3的耐久前及耐久后的铑平均粒径的图(实施例2)。图23是表示本发明的催化剂c-1及现有的催化剂c-2的耐久后的铂粒径分布的图(实施例3)。图24是表示本发明的催化剂d-1及现有的催化剂d-2的催化床温度的变化的图(实施例4〈评价1〉)。图25是表示本发明的催化装置x-1(a)及现有的催化装置x-2(b)的构成的图(实施例4〈评价2〉)。图26是表示使用本发明的催化装置x-1及现有的催化装置x-2时的排出气体的总碳氢化合物(thc)浓度的图(实施例4〈评价2〉)。具体实施方式〈废气净化催化剂〉本发明的废气净化催化剂具有结晶性金属氧化物载体及负载于该载体上的贵金属粒子,并且,贵金属粒子相对于结晶性金属氧化物载体即载体外延生长。特别是,在本发明的废气净化催化剂中,贵金属粒子在结晶性金属氧化物载体的结晶的平台(terrace)区域相对于该载体外延生长。根据本发明的废气净化催化剂,通过使贵金属粒子相对于结晶性金属氧化物载体外延生长,能够抑制催化剂使用期间的贵金属粒子的移动及由此导致的烧结。因此,本发明的废气净化催化剂即使在耐久以后,也能够维持较高的催化活性,例如,低温下的高催化活性。贵金属粒子在结晶性金属氧化物载体的结晶的平台区域(结晶表面的比较平坦的区域)相对于载体外延生长的状态为例如图1所示的状态。在此,图1中的4种圆圈(○)表示各个原子,在金属氧化物载体及贵金属粒子中,该圆圈整齐地排列,表明金属氧化物载体及贵金属粒子具有特定的结晶性。另外,金属氧化物载体和贵金属粒子能够形成同样的条纹,表明贵金属粒子相对于结晶性金属氧化物载体以同样的结晶取向而被负载,即,表明贵金属粒子相对于金属氧化物载体外延生长。与此相对,图2表示贵金属粒子并未相对于结晶性金属氧化物载体外延生长的状态,即,贵金属粒子并未相对于结晶性金属氧化物载体以同样的结晶取向而被负载的状态。在该图2所示的废气净化催化剂中,贵金属粒子负载于结晶的边缘区域及阶梯区域(形成单原子层的阶梯的区域)。关于本发明,贵金属粒子相对于载体外延生长的情况可通过透射电子显微镜(tem)的观察而得知。但是,关于本发明,贵金属粒子相对于载体外延生长,并非是指全部的贵金属粒子都相对于结晶性金属氧化物载体外延生长,而是指对于在用tem观察时能够判断是否进行外延生长的贵金属粒子的至少一部分、例如至少10%、至少30%、至少50%、至少70%、或实质上全部(tem照片的面积换算),贵金属粒子相对于结晶性金属氧化物载体外延生长。另外,在通过本发明的方法制造本发明的废气净化催化剂的情况下,关于本发明,贵金属粒子相对于载体外延生长是指,与不使用本发明的方法的情况相比,即,与不用掩蔽剂掩蔽载体的至少一部分表面的情况相比,贵金属粒子优先相对于载体外延生长,即,相对于载体外延生长的贵金属粒子的比例较大。在本发明的废气净化催化剂中,贵金属粒子不仅负载于载体的外表面,而且也分散负载于内表面。由此,与贵金属粒子仅负载于载体的外表面的情况相比,能够扩大贵金属粒子间的间隔,因此,能够进一步抑制贵金属粒子的烧结。在此,“载体的外表面”是指直接露出于载体的外侧的表面,另外,“载体的内表面”是指不直接露出于载体的外侧的表面,即,例如,细孔的内部和载体由二次粒子构成时的一次粒子间的间隙。〈废气净化催化剂-贵金属〉本发明的废气净化催化剂可使用的贵金属可以是任意的贵金属,考虑到废气净化性能,特别优选使用铂、钯、铑、或它们的组合。〈废气净化催化剂-载体〉本发明的废气净化催化剂可使用的载体可以是结晶性金属氧化物载体、尤其是粉末状的结晶性金属氧化物载体。作为这种载体,可列举例如选自由氧化铈、氧化锆、氧化铈-氧化锆固溶体、钙钛矿型金属氧化物、氧化钛及它们的组合构成的组的载体。这种金属氧化物载体优选为通过与负载于其上的贵金属的亲和性来吸附贵金属,由此抑制贵金属粒子移动的载体。因此,作为这种金属氧化物载体,优选使用如专利文献5举出的金属氧化物载体,即,优选使用构成金属氧化物载体的金属元素中的至少一种金属元素的阳离子具有比锆的阳离子小的电负性的载体。具体而言,作为这种载体,优选使用具有选自由如锂、钠、钾这样的碱金属;如钙、钡这样的碱土金属;如钇、铈这样的稀土类;以及它们的组合构成的组的金属元素的载体。特别是,作为这种金属氧化物载体,优选使用含有铈的金属氧化物载体,即,优选使用如氧化铈、或氧化铈-氧化锆复合氧化物这样的氧化铈系载体。在使用这种金属氧化物载体的情况下,如专利文献5所示,贵金属通过载体表面的氧与载体的阳离子键合,形成表面氧化物层,由此,能够抑制贵金属的粒子生长。〈废气净化催化剂-控制了贵金属粒子的形状及尺寸的方式〉本发明的废气净化催化剂的一个方式中,负载于载体上的贵金属粒子具有近似半球状的形状。在该方式中,通过使贵金属粒子以近似半球状的形态维持于载体上,载体和贵金属粒子的相接面积增大,由此能抑制催化剂使用期间的贵金属粒子的移动。具体而言,在该方式的废气净化催化剂中,如图5所示,载体和贵金属粒子相接的部分的宽度(w)比贵金属粒子距载体表面的高度(h)大,且满足下式(1):w/h>1.0、1.1、1.2、1.3、1.4、1.5或1.6…(1)。另外,在该方式的废气净化催化剂中,贵金属粒子距载体表面的高度(h)为0.5nm以上。即,在该方式的废气净化催化剂中,贵金属粒子并非以单独的原子的状态负载于载体上,而是以某种程度大小的粒子的形式而存在。这样,为了抑制催化剂使用期间贵金属粒子发生粒子生长到无法控制的程度、并且为了使贵金属以金属的状态存在于载体上,在废气净化催化剂中,优选使贵金属粒子以控制了尺寸的状态负载于载体上。与此相关,在本发明的废气净化催化剂中,贵金属粒子距载体表面的高度(h)可以为0.5nm以上、0.6nm以上、0.8nm以上、1.0nm以上、或2.0nm以上。另外,在本发明的废气净化催化剂中,贵金属粒子距载体表面的高度(h)可以为10.0nm以下、8.0nm以下、6.0nm以下、5.0nm以下、4.0nm以下、3.0nm以下、或2.0nm以下。需要说明的是,关于本发明,载体和贵金属粒子相接的部分的宽度(w)及贵金属粒子距载体表面的高度(h)是可通过透射电子显微镜(tem)观察而得到的值。但是,在本发明中,这些值并非全部贵金属粒子都满足,而是在用tem观察时能够测定这些值的贵金属粒子的至少一部分、例如至少10%、至少30%、至少50%、至少70%、或实质上全部(tem照片的面积换算)满足。如上所述,考虑到使用期间的贵金属粒子的烧结的抑制等,优选贵金属粒子以预定尺寸负载于载体上。这种贵金属粒子的尺寸也可以以通过一氧化碳吸附法测定的贵金属粒子的粒径来表示,通过一氧化碳吸附法测定的贵金属粒子的粒径优选为0.5~10.0nm,更优选为0.8~4.0nm,进一步优选为0.8~3.0nm。另外,这种贵金属粒子的粒度分布,例如,将下述的富气氛和贫气氛每2分钟切换一次,并在800℃下进行耐久5小时以后,可以具有3.0nm以下、2.0nm以下或1.5nm以下的半峰全宽,富气氛:co-3%、h2o-5%、n2-剩余部分,贫气氛:o2-5%、h2o-5%、n2-剩余部分。考虑到耐久后的废气净化催化剂的催化活性、尤其是低温时的催化活性,往往优选贵金属粒子在耐久以后具有较小的平均粒度,同时具有这种窄的粒度分布。〈废气净化催化剂-具体的方式〉根据上述的记载,本发明的废气净化催化剂例如可以为如下所述的催化剂:贵金属粒子以近似半球状的形状负载于载体上;载体与贵金属粒子相接的部分的宽度(w)和贵金属粒子距载体表面的高度(h)满足下式(1):w/h>1.0…(1);贵金属粒子距载体表面的高度(h)为0.5nm以上且10nm以下;贵金属粒子为铂;且载体为氧化铈或氧化铈-氧化锆固溶体。另外,根据上述的记载,本发明的废气净化催化剂例如可以为如下所述的催化剂:贵金属粒子以近似半球状的形状负载于载体上;载体与贵金属粒子相接的部分的宽度(w)和贵金属粒子距载体表面的高度(h)满足下式(1):w/h>1.0…(1);贵金属粒子距载体表面的高度(h)为2nm以上且5nm以下;贵金属粒子为铑;且载体为氧化铈或氧化铈-氧化锆固溶体。在此,贵金属粒子和载体也可以具有(111)面之间或(200)面之间的晶面接合。〈废气净化催化剂-制造方法〉这种本发明的废气净化催化剂可通过任意方法来制造,特别是,可通过制造废气净化催化剂的本发明的方法来制造。〈废气净化装置〉本发明的废气净化装置具有基材及保持在基材上的废气净化催化剂,并且,保持在基材上的废气净化催化剂中的、在基材的废气流上游侧负载的废气净化催化剂是本发明的废气净化催化剂。作为本发明的废气净化装置所使用的基材,可以列举蜂窝基材,例如,董青石制蜂窝基材。作为在本发明的废气净化装置的基材的废气流下游侧负载的废气净化催化剂,可使用任意的其他废气净化催化剂。另外,本发明的废气净化装置的基材能够在直到基材的废气流上游侧的三分之二的区域、直到二分之一的区域、或直到三分之一的区域保持本发明的废气净化催化剂。〈废气净化催化剂的制造方法〉制造废气净化催化剂的本发明的方法包括如下工序:在溶液中,利用掩蔽剂将载体的至少一部分表面掩蔽;且将由掩蔽剂掩盖了至少一部分表面的载体导入含有贵金属的含贵金属溶液中;然后,对载体及含贵金属溶液进行干燥及煅烧,使贵金属负载于载体上。通常,在将结晶性金属氧化物载体导入含贵金属溶液的情况下,贵金属具有吸附或配位于结晶性金属氧化物载体的缺陷部分、例如结晶的边缘区域、阶梯区域(形成单原子层的阶梯的区域)、扭折区域(阶梯扭曲的区域)等,从而负载于这些缺陷部分的倾向。与此相对,根据该本发明的方法,预先由掩蔽剂掩蔽结晶性金属氧化物载体的至少一部分表面。在此,掩蔽剂与贵金属同样,具有吸附或配位于结晶的缺陷部分的倾向。因此,当将这种被掩蔽后的载体导入含贵金属溶液时,贵金属并不会吸附或配位于已被掩蔽剂掩蔽的结晶的缺陷部分,取而代之的是吸附或配位于结晶的没有缺陷的部分、即结晶的平台区域等。因此,当将使贵金属吸附或配位后的载体干燥和煅烧时,较多的贵金属负载于载体的平台区域等结晶缺陷少的区域。另外,掩蔽剂通过该煅烧而被燃烧除去。负载于载体的结晶缺陷少的区域的贵金属粒子易受载体的结晶性的影响。因此,通过煅烧等,能够使贵金属粒子相对于载体外延生长,即,能够使贵金属粒子的结晶取向与负载有贵金属粒子的结晶性金属氧化物载体相同。〈废气净化催化剂的制造方法-掩蔽剂〉本发明的方法中使用的该掩蔽剂,可以是能够掩蔽结晶性金属氧化物载体的结晶缺陷的任意的掩蔽剂。作为这种掩蔽剂,可以列举具有能够配位于结晶性金属氧化物载体的官能团、例如选自由仲氨基、叔氨基、羧基、羟基、羰基及它们的组合构成的组的官能团的化合物。因此,作为具体的掩蔽剂,可以列举具有一个或多个上述官能团的有机化合物,特别而言,可以列举多元芳族羧酸,例如:对苯二甲酸、间苯二甲酸、均苯三甲酸及均苯四甲酸。〈废气净化催化剂的制造方法-载体及贵金属〉作为制造废气净化催化剂的本发明的方法中可使用的载体及贵金属,可参照本发明的废气净化催化剂相关的上述的记载。作为用于负载贵金属的贵金属溶液,可以使用如二硝基二氨络合物硝酸溶液、氯铂酸溶液这样的在负载贵金属时通常使用的贵金属溶液、可以使用贵金属胶体、可以使用下述所示的部分地还原和凝聚的贵金属凝聚体等。在能够控制负载于载体的贵金属粒子的粒径方面、能够改良载体和负载于载体的贵金属粒子的密接性且促进贵金属粒子的外延生长的方面,优选使用下述所示的部分地还原和凝聚的贵金属凝聚体。另外,在使用通过与负载于其上的贵金属的亲和性来吸附贵金属的金属氧化物载体作为载体的情况下,在通过干燥及煅烧将部分地还原和凝聚而成的贵金属凝聚体从含贵金属凝聚体溶液负载到载体上时,通过载体和贵金属的亲和性,吸附贵金属,由此,能够促进与载体较密接的形态的贵金属粒子的生成。〈废气净化催化剂的制造方法-控制贵金属粒子的形状及尺寸的方式〉对于制造废气净化催化剂的本发明的方法的一个方式而言,含贵金属溶液是含有部分地还原和凝聚而成的贵金属凝聚体的含贵金属凝聚体溶液。在此,该含贵金属凝聚体溶液可通过如下方式来得到:提供含有贵金属离子和/或配位化合物和聚合物的原料溶液,使贵金属离子和/或配位化合物配位于聚合物,然后,使配位于聚合物的贵金属离子和/或配位化合物部分地还原和凝聚。另外,关于本发明,贵金属离子和/或配位化合物部分地还原和凝聚是指,通过加热、还原剂的供给等来进行还原和凝聚,直到能够使贵金属离子和/或配位化合物进一步还原和/或凝聚的状态。在此,贵金属离子和/或配位化合物部分地还原和凝聚可通过例如对含有这种贵金属凝聚体的溶液进行吸光度测定来判断。即,吸光度可以表示贵金属粒子的凝聚程度,吸光度大表示在含贵金属凝聚体溶液中,贵金属形成了大的凝聚体和/或贵金属形成了密集的凝聚体,因此贵金属进行了“部分地还原和凝聚”的状态是指能够进一步进行“还原和凝聚”的状态,即,能够进一步增大吸光度的状态。下面,利用图7对该方式的原理进行概念性地说明,但本发明并不受这些说明的任何限定。如图7所示,在该方式中,首先,提供含有贵金属离子和/或配位化合物的贵金属溶液(41)及含有聚合物的聚合物溶液(42),将这两种溶液混合(21),使贵金属离子和/或配位化合物配位于聚合物(43)。使该配位于聚合物的贵金属离子和/或配位化合物部分地还原和凝聚(22),在聚合物中形成部分地还原和凝聚而成的贵金属凝聚体(44)。其后,将由掩蔽剂掩蔽了一部分表面的载体(35)导入含有该凝聚体的溶液中,然后,对载体及贵金属凝聚体进行干燥及煅烧(23、24),使贵金属凝聚体(31)负载于载体(35)上。在该方式中,将仅进行了部分地还原和凝聚的贵金属凝聚体、即尚未完全还原和凝聚而较松散地凝聚的贵金属凝聚体负载于载体。由此,能够使贵金属粒子以与载体比较密接的方式,以近似半球状的形态负载于载体上。这样,贵金属粒子和载体比较密接,由此,在贵金属粒子以近似半球状的形态负载于载体上的情况下,贵金属粒子和载体的相接面积大,由此能够抑制催化剂使用期间的贵金属移动及由该移动造成的贵金属的粒子生长。进而,在该方式中,通过使贵金属以预定尺寸的粒子存在,能够促进催化活性。与此相对,在通过含有贵金属离子和/或配位化合物的贵金属溶液(41)而将贵金属直接负载于载体上的情况(25)、及将贵金属溶液和聚合物溶液混合以后(43)不进行贵金属离子和/或配位化合物的部分还原和凝聚就将贵金属负载于载体上的情况(26)下,贵金属以原子水平分散而负载于载体上(32),因此,无法得到贵金属以预定尺寸的粒子存在所带来的益处。另外,在使配位于聚合物的贵金属离子或配位化合物高度还原和凝聚(28)的情况下,在聚合物中,贵金属原子之间不断凝聚,最终形成球状的稳定结构(45)。在将载体(35)导入含有这种凝聚体的溶液中,然后对载体及原料溶液进行干燥及煅烧而使贵金属凝聚体负载于载体的情况(27)下,贵金属牢固地凝聚而具有球状的稳定结构(33),不能使其相对于载体密接。因此,在这种情况下,贵金属和载体的相接面积小,因此难以抑制催化剂使用期间的贵金属的移动及由该移动造成的贵金属的粒子生长。另外,在该方式中,由于在溶液中分散有贵金属凝聚体,且将载体导入该溶液中使贵金属粒子负载于载体上,因此不仅能够使贵金属凝聚体负载于载体的外表面,而且也能够使其负载于载体的内表面、例如细孔的内部、载体由二次粒子构成时的一次粒子间的间隙等。〈废气净化催化剂的制造方法-控制贵金属粒子的形状及尺寸的方式-部分还原和凝聚〉在控制贵金属粒子的形状及尺寸来制造废气净化催化剂的方式中,能够用任意方法使配位于聚合物的贵金属离子和/或配位化合物部分地还原和凝聚。作为进行部分还原和凝聚的具体方法,可以列举如加热回流这样的加热处理、还原剂的添加、以及它们的组合等。在通过加热处理进行还原和凝聚的情况下,通过控制加热程度和/或加热时间,能够实现贵金属离子和/或配位化合物的部分还原和凝聚。另外,在使用还原剂进行还原和凝聚的情况下,通过还原剂种类的选择、还原剂使用量的调节等,能够实现贵金属离子和/或配位化合物的部分还原和凝聚。作为还原剂,可以列举例如如甲醇、乙醇、丙醇这样的醇。另外,贵金属离子和/或配位化合物的部分还原和凝聚的程度可依据期望的贵金属粒子的尺寸、贵金属粒子的形状等来确定。通常,贵金属离子和/或配位化合物的部分还原和凝聚的程度小,因此,在贵金属比较松散地凝聚的情况下,具有贵金属粒子的尺寸减小和/或贵金属粒子成为与载体密接的形状的倾向。〈废气净化催化剂的制造方法-控制贵金属粒子的形状及尺寸的方式-聚合物〉作为控制贵金属粒子的形状及尺寸来制造废气净化催化剂的方式中可以使用的聚合物,可使用能够使贵金属离子和/或配位化合物配位的任意聚合物。因此,作为这种聚合物,可以使用例如具有仲氨基、叔氨基、羧基、羟基、羰基等能够使金属配位的官能团的聚合物,特别是具有仲氨基、叔氨基等官能团的聚合物,例如,聚乙烯吡咯烷酮。另外,作为本发明方法中可以使用的聚合物,也可以使用如专利文献1及2所使用的树枝状聚合物。本发明方法中使用的聚合物的使用量可依据聚合物使贵金属离子和/或配位化合物配位的能力、期望的贵金属粒子的尺寸、期望的贵金属粒子的形状等而任意地确定。实施例《实施例1》〈铂负载氧化铈-氧化锆催化剂a-1〉向铂的总含量为4.50×10-3mol的二硝基二氨铂硝酸([pt(no2)2(nh3)2])溶液中添加离子交换水并进行搅拌,得到300g的稀释铂溶液。向以单体单位换算计2.25×10-2mol(铂的摩尔数的5倍)的聚乙烯吡咯烷酮(pvp)2.52g中添加300g离子交换水并进行搅拌,使聚乙烯吡咯烷酮完全溶解,制备均匀的聚乙烯吡咯烷酮溶液。其后,向聚乙烯吡咯烷酮溶液中缓慢地滴入稀释铂溶液进行混合,在室温下搅拌1小时,然后添加作为还原剂的乙醇,以使离子交换水与乙醇的混合比率为20:80(质量比),然后,搅拌30分钟,得到铂-聚乙烯吡咯烷酮溶液。将这样得到的铂-聚乙烯吡咯烷酮溶液加热回流,使铂离子还原,得到含铂微粒溶液。在此,加热回流在100℃下进行4小时,得到含铂微粒溶液。使30g的氧化铈-氧化锆固溶体(结晶性,ce:zr=7:3,用共沉淀法制成)的载体粉末分散于180g的蒸馏水中,向该溶液中添加60g均苯四甲酸并进行搅拌,其后,添加通过进行加热回流而得到的上述铂微粒溶液,以使铂相对于载体粉末为0.5重量%,搅拌1小时。其后,在120℃下使水分蒸发,在500℃下进行2小时的煅烧,用研钵进行粉碎,制备铂负载氧化铈-氧化锆催化剂a-1。〈铂负载氧化铈-氧化锆催化剂a-2〉除未添加均苯四甲酸以外,与铂负载氧化铈-氧化锆催化剂a-1同样地操作,制备铂负载氧化铈-氧化锆催化剂a-2。〈铂负载氧化铈-氧化锆催化剂a-3〉直接使用二硝基二氨铂硝酸溶液而并不制备含铂微粒溶液,除此以外,与铂负载氧化铈-氧化锆催化剂a-1同样地操作,制备铂负载氧化铈-氧化锆催化剂a-3。〈观察〉对于该催化剂a-1及a-2,进行透射电子显微镜(tem)观察。将结果示出于图3。图3(a)是使用均苯四甲酸作为掩蔽剂的催化剂a-1的tem照片,另外,图3(b)是未使用掩蔽剂的催化剂a-2的tem照片。由图3(a)和图3(b)的比较可知,在作为掩蔽剂而使用均苯四甲酸的催化剂a-1(图3(a))中,铂粒子在结晶性金属氧化物载体的平台区域外延生长,与此相对,在不使用掩蔽剂的催化剂a-2(图3(b))中,铂粒子负载于结晶性金属氧化物载体的边缘区域。对于可用tem观察到这些值的其他贵金属粒子,也观察到了同样的倾向。〈评价〉对于铂负载氧化铈-氧化锆催化剂a-1~a-3,成形为颗粒状,利用图13所示的装置,对排出气体的碳氢化合物(hc)浓度如下进行评价。耐久条件:将富气氛及贫气氛每2分钟切换一次,并在900℃下加热5小时。废气净化催化剂量:2.0g(铂浓度:0.5质量%)评价条件:氮气气氛化且升温到500℃,在氢气1.0%的条件下,还原5分钟。其后,切换到评价气体条件(c3h6:3000ppmc、o2:10%、co2:12%、h2o:5%),以10℃/分钟的速度降温,用连续分析仪测定该排出气体。气体流量为20l/分钟。将评价结果示出于图4。由图4可知,未制备含铂微粒溶液而直接使用二硝基二氨铂硝酸溶液的催化剂a-3的性能最差。另外,与不使用掩蔽剂的催化剂a-2相比,使用均苯四甲酸作为掩蔽剂的催化剂a-1具有优良的碳氢化合物(hc)净化性能。需要说明的是,催化剂a-1~a-3的碳氢化合物(hc)50%净化温度(hc的净化率为50%的温度)分别为155℃、184℃及223℃。《实施例2》〈铑负载氧化铈催化剂b-1〉向铑的总含量为7.50×10-3mol的硝酸铑{rh(no3)3}溶液中添加离子交换水并进行搅拌,得到300g的稀释铑溶液。向以单体单位换算计2.25×10-2mol(铑的摩尔数的3倍)的聚乙烯吡咯烷酮(pvp)2.52g中添加300g离子交换水并进行搅拌,使聚乙烯吡咯烷酮完全溶解,制备均匀的聚乙烯吡咯烷酮溶液。然后,向聚乙烯吡咯烷酮溶液中缓慢地滴入稀释铑溶液并进行混合,在室温下搅拌1小时,添加作为还原剂的1-丙醇,以使离子交换水与1-丙醇的混合比率为20:80(质量比),然后,搅拌30分钟,得到铑-聚乙烯吡咯烷酮溶液。将这样得到的铑-聚乙烯吡咯烷酮溶液加热回流,使铑离子还原,得到含铑微粒溶液。在此,加热回流在100℃下进行4小时,得到含铑微粒溶液。使10g的氧化铈(预先在大气中、1000℃下进行5小时的煅烧)的载体粉末分散于60g的蒸馏水中,向该溶液中添加20g均苯四甲酸并进行搅拌,其后,添加进行加热回流而得到的上述铑微粒溶液,以使铑相对于载体粉末为1.0重量%,搅拌1小时。其后,在120℃下使水分蒸发,在450℃下进行2小时的煅烧,用研钵进行粉碎,制备铑负载氧化铈催化剂b-1。〈铑负载氧化铈催化剂b-2〉使用氯化铑溶液代替硝酸铑溶液、使用乙醇代替1-丙醇、并且在80℃下进行4小时的加热回流,除此以外,与铑负载氧化铈催化剂b-1同样地操作,制备铑负载氧化铈催化剂b-2。〈铑负载氧化铈催化剂b-3〉直接使用硝酸铑溶液而并不制备含铑微粒溶液,除此以外,与铑负载氧化铈催化剂b-1同样地操作,制备铑负载氧化铈催化剂b-3。〈观察〉将对催化剂b-1进行透射电子显微镜(tem)观察的结果示出于图17~19。在图17中,在载体上存在5nm以下的铑粒子的部位,可观察到莫尔条纹,并且,观察到该莫尔条纹大多朝向同一方向。表明这些莫尔条纹与载体的结晶方向和铑的结晶方向有关。另外,图18及19示出对存在于载体的端面的各个铑粒子进行观察的结果。由该图18及19可知,铑及载体具有(111)面之间或(200)面之间的晶面接合。将对催化剂b-2进行透射电子显微镜(tem)观察的结果示出于图20。由图20可知,在载体上存在大小超过约5nm的铑粒子的部位(用箭头表示)没有观察到莫尔条纹。这表明,大小超过5nm的铑粒子的结晶方向与载体的结晶方向无关或相关性小。将对催化剂b-3进行透射电子显微镜(tem)观察的结果示出于图21。由图21可知,在直接使用硝酸铑溶液将铑负载于载体的情况下,载体上的铑粒子的粒径偏差较大,并且,未观察到莫尔条纹。〈评价〉对于铑负载氧化铈催化剂b-1及b-3,如下对热耐久前后铑的平均粒径进行评价。耐久条件:将富气氛及贫气氛每2分钟切换一次,并在1000℃下加热5小时。试样:0.1g一氧化碳吸附条件:将废气净化催化剂在氧气中、400℃下加热20分钟来进行氧化处理,然后,在氢气中、400℃下加热20分钟来进行还原处理,其后,在0℃下,使一氧化碳吸附于废气净化催化剂。将评价结果示出于图22。图22显示,与通过使用硝酸铑直接负载铑的方法而得到的现有的铑负载氧化铈催化剂b-3相比,本发明的铑负载氧化铈催化剂b-1耐久后的铑粒径维持在较小水平。认为这是因为,对于本发明的催化剂b-1而言,由于载体和铑粒子之间的相互作用大,载体上的贵金属粒子的移动得到抑制,由此抑制了贵金属粒子的结块。《实施例3》〈铂负载氧化铈-氧化锆催化剂c-1〉与铂负载氧化铈-氧化锆催化剂a-1同样地操作,使用聚乙烯吡咯烷酮溶液及稀释铂溶液,制备本发明的铂负载氧化铈-氧化锆催化剂c-1。〈铂负载氧化铈-氧化锆催化剂c-2〉直接使用二硝基二氨铂硝酸溶液而并不制备含铂微粒溶液,除此以外,与铂负载氧化铈-氧化锆催化剂c-1同样地操作,制备现有的铂负载氧化铈-氧化锆催化剂c-2。〈评价〉对于铂负载氧化铈-氧化锆催化剂c-1及c-2,如下对热耐久后铂的平均粒径分布进行评价。耐久条件:将下述的富气氛及贫气氛每2分钟切换一次,在800℃下加热5小时。富气氛:co-3%、h2o-5%、n2-剩余部分,贫气氛:o2-5%、h2o-5%、n2-剩余部分。评价条件:通过酸来溶解氧化铈-氧化锆载体,得到仅分散有铂粒子的铂粒子分散液。其后,使用动态光散射法粒径分布分析装置(dls)对该铂粒子分散液进行分析,得到铂粒子的粒度分布。需要说明的是,由于铂粒子不受酸的影响,因此通过该方法,能够得到铂负载氧化铈-氧化锆催化剂的铂粒子的粒度分布。将评价结果示出于图23。另外,作为参考,关于铂负载氧化铈-氧化锆催化剂c-2,将耐久前的粒径分布也示出于图23。另外,对于用聚乙烯吡咯烷酮溶液及稀释铂溶液制备的铂负载氧化铈-氧化锆催化剂c-1而言,耐久后的平均粒度为约2.6nm。另外,对于未制备含铂微粒溶液而直接使用二硝基二氨铂硝酸溶液的催化剂c-2而言,耐久后的平均粒度为约2.4nm。由图23可知,对于用聚乙烯吡咯烷酮溶液及稀释铂溶液制备的铂负载氧化铈-氧化锆催化剂c-1而言,耐久后的粒度分布窄,半峰全宽为约1nm。认为这是因为,对于催化剂c-1而言,在其制备时,贵金属以比较均匀的大小负载于载体上,并且载体与贵金属的相互作用大,由此抑制了耐久期间的贵金属的结块。与此相对,由图23可知,对于未制备含铂微粒溶液而直接使用二硝基二氨铂硝酸溶液的催化剂c-2而言,耐久前,平均粒度小,且粒度也均匀,与此相对,耐久后,粒度分布变大,半峰全宽超过4nm。认为这是因为,对于催化剂c-2而言,在其制备时,虽然贵金属以非常微细的大小负载于载体上,但载体与贵金属的相互作用小,由此在耐久期间发生贵金属的结块。《实施例4》〈铂负载氧化铈-氧化锆催化剂d-1〉与铂负载氧化铈-氧化锆催化剂a-1同样地操作,使用聚乙烯吡咯烷酮溶液及稀释铂溶液,制备本发明的铂负载氧化铈-氧化锆催化剂d-1。〈铂负载氧化铈-氧化锆催化剂d-2〉直接使用二硝基二氨铂硝酸溶液而并不制备含铂微粒溶液,除此以外,与铂负载氧化铈-氧化锆催化剂d-1同样地操作,制备现有的铂负载氧化铈-氧化锆催化剂d-2。〈评价1〉关于铂负载氧化铈-氧化锆催化剂d-1及d-2,成形为颗粒状,用图13所示的装置,如下对催化床温度的速度进行评价。耐久条件:将富气氛及贫气氛每2分钟切换一次,在900℃下加热5小时。废气净化催化剂量:2.0g(铂浓度:0.5质量%)评价条件:氮气气氛化且升温到500℃,在氢气1.0%的条件下,还原5分钟。其后,切换到评价气体条件(c3h6:3000ppmc、o2:10%、co2:12%、h2o:5%),使吸入气体温度以10℃/分钟的速度升温,测定催化床温度。气体流量为20l/分钟。将评价结果示出于图24。由图24可知,与现有的铂负载氧化铈-氧化锆催化剂即催化剂c-2相比,本发明的铂负载氧化铈-氧化锆催化剂c-1,虽然初期的催化床温度上升较慢,但催化床温度的上升一旦开始,就迅速地进行直到实现优选的废气净化率的约400℃的催化床温度的上升。认为这是因为,对于本发明的铂负载氧化铈-氧化锆催化剂c-1而言,如实施例3所示,贵金属的粒度分布窄,由此在特定的温度下,使多数贵金属粒子一次性地活化。与此相对,对于现有的铂负载氧化铈-氧化锆催化剂c-2而言,如实施例3所示,贵金属的粒度分布广,虽然也存在非常微细的贵金属粒子,但所述微细的贵金属粒子相对于整体的比例小,因此为了使较大比例的贵金属粒子活化,需要比较高的温度。〈评价2〉如图25(a)所示,将铂负载氧化铈-氧化锆催化剂d-1及d-2负载于董青石制蜂窝载体上,即,将铂负载氧化铈-氧化锆催化剂d-1自蜂窝载体的废气入口侧涂敷到其三分之一处,并且,将铂负载氧化铈-氧化锆催化剂d-2自蜂窝载体的废气出口侧涂敷到其三分之二处,得到本发明的催化装置x-1。如图25(b)所示,将铂负载氧化铈-氧化锆催化剂d-2负载于董青石制蜂窝载体上,即,在蜂窝状载体的整体上涂敷铂负载氧化铈-氧化锆催化剂d-2,得到比较例的催化装置x-2。对于本发明的催化装置x-1及现有的催化装置x-2,如下对排出气体的总碳氢化合物(thc)浓度进行评价。耐久条件:使排气量为2.4l的发动机以高负荷运转,供给其废气,在催化床温度900℃下加热50小时。废气净化催化剂量:2.0g(铂浓度:0.5质量%)评价条件:将本发明的催化装置x-1及比较例的催化装置x-2分别与实际的发动机连接,用连续分析仪测定其排出气体。气体流量为20l/分钟。将评价结果示出于图26。由图26可知,与比较例的催化装置x-2相比,本发明的催化装置x-1,在比较早的阶段就出现排出气体的碳氢化合物浓度降低,即,催化装置的起动性能优良。认为这是因为,与现有的铂负载氧化铈-氧化锆催化剂d-2相比,本发明的铂负载氧化铈-氧化锆催化剂d-1,虽然初期的催化床温度上升较慢,但催化床温度的上升一旦开始,其后催化床温度就迅速上升。《参考例1》向铂的总含量为4.50×10-3mol的二硝基二氨铂硝酸([pt(no2)2(nh3)2])溶液中添加离子交换水并进行搅拌,得到300g的稀释铂溶液。向以单体单位换算计2.25×10-2mol(铂的摩尔数的5倍)的聚乙烯吡咯烷酮(pvp)2.52g中添加300g离子交换水并进行搅拌,使聚乙烯吡咯烷酮完全溶解,制备均匀的聚乙烯吡咯烷酮溶液。然后,向聚乙烯吡咯烷酮溶液中缓慢地滴入稀释铂溶液并进行混合,在室温下搅拌1小时,添加乙醇,以使离子交换水与乙醇的混合比率为20:80(质量比),然后搅拌30分钟,得到铂-聚乙烯吡咯烷酮溶液。将这样得到的铂-聚乙烯吡咯烷酮溶液加热回流,使铂离子还原,得到含铂微粒溶液。在此,加热回流在100℃下,进行1小时、4小时及8小时,得到三种含铂微粒溶液。将30g的氧化铈(ceo2)的载体粉末分散于180g的蒸馏水中,并向其中分别添加未进行加热回流的铂-聚乙烯吡咯烷酮溶液及进行加热回流而得到的上述三种铂微粒溶液,以使铂相对于载体粉末为0.5重量%,搅拌1小时。其后,在120℃下使水分蒸发,在450℃下进行2小时的煅烧,用研钵进行粉碎,制备四种铂负载氧化铈催化剂。另外,同样地操作,使用进行了4小时的加热回流的含铂微粒溶液,将铂负载于氧化铝载体粉末上,制备铂负载氧化铝催化剂。对于这些铂负载氧化铈催化剂,进行透射电子显微镜(tem)观察。将结果示出于图8~11。图8~10分别是使用进行1小时、4小时及8小时的加热回流而得到的含铂微粒溶液使铂负载于氧化铈载体粉末的铂负载氧化铈催化剂(pt/ceo2)。另外,图11是使用进行4小时的加热回流而得到的含铂微粒溶液使铂负载于氧化铝载体粉末的铂负载氧化铝催化剂(pt/al2o3)。另外,对于使用未进行加热回流的铂-聚乙烯吡咯烷酮溶液而得到的铂负载氧化铈催化剂(pt/ceo2)而言,铂以非常微细的状态负载于氧化铈载体粉末,难以用tem观察到铂粒子。关于上述的铂负载氧化铈催化剂(pt/ceo2)及铂负载氧化铝催化剂(pt/al2o3)的评价结果,汇总于下述的表1。[表1]催化剂构成加热回流时间w(nm)h(nm)w/hpt/ceo2无---pt/ceo2(图4)1小时2.281.361.68pt/ceo2(图5)4小时1.441.041.38pt/ceo2(图6)8小时1.441.680.86pt/al2o3(图7)4小时--0.75由上述的表1可知,在使用通过加热回流使铂部分地还原和凝聚而成的含铂微粒溶液来得到铂负载氧化铈催化剂的情况下,即,在进行了2小时或4小时的加热回流的情况下,氧化铈载体与铂粒子相接的部分的宽度(w)比铂粒子距氧化铈载体表面的高度(h)长。对于实质上全部的用tem能够观察这些值的贵金属粒子,观察到了同样的倾向。另外,由图8和图11的比较可知,即使在使用进行4小时的加热回流而得到的含铂微粒溶液的情况下,负载于氧化铈载体上的铂粒子也为半球状,负载于氧化铝载体的铂粒子为近似于球状的形状。认为这是因为,氧化铈载体相对于贵金属显示较强的亲和性,与此相对,氧化铝载体相对于贵金属并不具有较强的亲和性。因此,贵金属离子或配位化合物的还原和凝聚的程度需要依赖于使用的载体来进行调节。《参考例2》将如参考例1那样得到的聚乙烯吡咯烷酮-铂溶液加热回流,使铂离子还原和凝聚,得到含铂微粒溶液。在此,加热回流进行1小时、2小时、4小时及8小时,得到四种含铂微粒溶液。对于如参考例1那样得到的聚乙烯吡咯烷酮-铂溶液及进行了加热回流的四种含铂微粒溶液,测定220nm~780nm的波长下的吸光度。将结果示出于图12。需要说明的是,该吸光度能够显示铂粒子的凝聚程度,吸光度大表明在聚乙烯吡咯烷酮-铂溶液及含铂微粒溶液中,铂形成较大的凝聚体和/或铂形成密集的凝聚体。另外,在将入射光强度设为i0,并且将透射光强度设为i时,吸光度为用log10(i0/i)表示的物体的光吸收强度。如参考例1那样得到的聚乙烯吡咯烷酮-铂溶液(“未加热回流”)的最大吸光度为约2.7。另外,进行了1小时、2小时、4小时及8小时的加热环流即还原和凝聚处理的四种含铂微粒溶液的最大吸光度分别为约2.9、约3.3、约3.3及约3.5。由该结果可知,在直到至少4小时为止的加热回流下,铂离子的还原和凝聚未完全进行,通过继续加热回流,铂离子的还原和凝聚会进一步进行。即,由该结果可知,在直到至少4小时为止的加热回流下,铂离子的还原和凝聚仅部分地进行。《参考例3》使用如参考例1那样进行了4小时的加热回流而得到的含铂微粒溶液,使铂负载于氧化铈载体,得到铂负载氧化铈催化剂3-1(pt/ceo2)。另外,直接使用如参考例1那样得到的稀释铂溶液、即稀释二硝基二氨铂硝酸([pt(no2)2(nh3)2])溶液代替含铂微粒溶液,除此以外,与参考例1同样地操作,使铂负载于氧化铈载体粉末,得到铂负载氧化铈催化剂3-2(pt/ceo2)。对于这些本发明及现有技术的废气净化催化剂,利用如图13所示的装置,用下述所示的一氧化碳(co)脉冲吸附法求出耐久后的铂粒径。耐久条件:将富气氛及贫气氛每2分钟切换一次,在1000℃下加热5小时。试样:0.1g一氧化碳吸附条件:将废气净化催化剂在氧气中、400℃下加热20分钟来进行氧化处理,然后,在氢气中、400℃下加热20分钟来进行还原处理,其后,在0℃下,使一氧化碳吸附于废气净化催化剂。将所测定的耐久后的铂粒径示出于图14。由图14可知,与废气净化催化剂3-2相比,废气净化催化剂3-1在耐久后也能够维持较小的铂粒径。《参考例4》〈废气净化催化剂4-1的制造〉如参考例1那样向铂的总含量为4.50×10-3mol的二硝基二氨铂硝酸([pt(no2)2(nh3)2])溶液中添加离子交换水并进行搅拌,得到300g的稀释铂溶液。向以单体单位换算计2.25×10-2mol(铂的摩尔数的5倍)的聚乙烯吡咯烷酮(pvp)2.52g中添加300g离子交换水并进行搅拌,使聚乙烯吡咯烷酮完全溶解,制备均匀的聚乙烯吡咯烷酮溶液。然后,向聚乙烯吡咯烷酮溶液中缓慢地滴入稀释铂溶液并进行混合,在室温下搅拌1小时,添加乙醇,以使离子交换水与乙醇的混合比率为20:80(质量比),然后,搅拌30分钟,得到铂-聚乙烯吡咯烷酮溶液。将这样得到的铂-聚乙烯吡咯烷酮溶液进行2小时的加热回流,使铂离子还原和凝聚,得到含铂微粒溶液。将这样得到的含铂微粒溶液作为含铂微粒溶液4-1。将30g的氧化铈(ceo2)系氧化物的粉末分散到以重量计6倍的蒸馏水中,向其中添加含铂微粒溶液4-1,以使pt相对于粉末为0.5重量%,搅拌1小时。其后,在120℃下使水分蒸发,在450℃下进行2小时的煅烧,用研钵进行粉碎,制备铂负载氧化铈催化剂。将这样制备的铂负载氧化铈催化剂作为废气净化催化剂4-1。该废气净化催化剂4-1与在参考例1中进行2小时的加热回流而得到的废气净化催化剂相对应。将废气净化催化剂4-1的制造条件及分析结果汇总于下述的表2中。另外,关于吸光度及铂粒径,分别如参考例2及3那样进行测定。〈废气净化催化剂4-2~4-8的制造〉改变下述的表2所示的制造条件,除此以外,与废气净化催化剂4-1同样地操作,得到废气净化催化剂4-2~4-8。将废气净化催化剂4-2~4-8的制造条件及分析结果汇总于下述的表2中。[表2]铂微粒溶液的制造条件及分析结果*1pvp(聚乙烯吡咯烷酮)以单体单位换算〈评价〉关于废气净化催化剂4-1~4-8,成形为颗粒状,利用如图13所示的装置,如下对碳氢化合物(hc)50%净化温度(hc的净化率达到50%的温度)进行评价。气体总流量:15l/分钟气体组成:c3h6-1000ppm或3000ppmc、co-6500ppm、no-1500ppm、o2-7000ppm、co2-10%、h2o-无、n2-剩余部分温度条件:从600℃降温至100℃(降温速度为20℃/分钟)废气净化催化剂量:3.0g将评价结果示出于图15。由图15可知,在铂的平均粒径为0.8~4nm(与铂原子数为9~1140个的粒子相对应)时,hc50%净化温度低,即,废气净化催化剂的活性高。这是因为,在铂的粒径过小时,铂以接近氧化物的状态存在,在铂的粒径过大时,作为反应的活性部位的铂的表面积减小。《参考例5》与参考例4的废气净化催化剂4-1及4-4同样地操作,得到废气净化催化剂5-1及5-2。在此,废气净化催化剂5-1及5-2的平均铂粒径分别为1.4nm及0.7nm。对于这样得到的废气净化催化剂5-1及5-2,利用x射线光电子能谱(xps)分析,由4f7/2轨道的结合能,对废气净化催化剂5-1(平均铂粒径1.4nm)及废气净化催化剂5-2(平均铂粒径0.7nm)中的铂的氧化状态进行评价。将结果示出于图16。由该图16可知,在废气净化催化剂5-1(平均铂粒径1.4nm)中,铂为更倾向金属的状态(pt0),与此相对,在废气净化催化剂5-2(平均铂粒径0.7nm)中,铂为更倾向氧化物的状态(pt2+)。标号说明10、10a、10b、10c废气净化催化剂11载体12、12a、12b、12c贵金属粒子w载体与贵金属粒子相接的部分的宽度h贵金属粒子距载体表面的高度31、32、33贵金属粒子35载体70参考例3及4所使用的试验装置当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1