一种高分散吸附脱硫催化剂及其制备和应用的制作方法
本发明属于石油化工领域,具体涉及一种活性组分高分散的吸附脱硫催化剂及其制备方法,以及在超深度吸附脱硫反应中的应用。
背景技术:
近年来,全球气候环境恶化,雾霾天气频现,快速增加的机动车辆和含硫尾气的排放是雾霾天气的主要诱因之一。日趋严格的环境法规极大地推动了全球车用汽油的质量升级进程,欧v标准已于2009年在欧洲全面推行(规定汽柴油硫含量低于10ppm),我国也已经于2018年底要求国v汽油标准在全国范围实施。因此,发展超低硫清洁油品的生产是当下迫在眉睫的任务。
目前,工业上广泛采用的汽油脱硫技术主要有加氢脱硫工艺和吸附脱硫工艺。采用加氢脱硫工艺在深度脱除汽油中的含硫化合物时,由于不可避免地会发生烯烃组分的饱和副反应,而汽油中的烯烃组分对其辛烷值有很大贡献,因此导致了一定的辛烷值损失。相比之下,吸附脱硫工艺由于吸附剂不含高加氢活性的组分,因此其脱硫选择性高、产品辛烷值损失小。此外,使用固定床工艺进行汽油的吸附脱硫还具有工艺简单、操作条件温和和脱硫深度高等特点。
吸附脱硫工艺的原理是通过吸附脱硫催化剂对汽油中含硫化合物的吸附降低汽油产品的硫含量,根据具体吸附原理又可分为物理吸附脱硫和催化反应吸附脱硫。其中物理吸附脱硫过程不涉及含硫化合物分子的化学反应,仅通过吸附剂上吸附位点与含硫化合物分子的相互作用将其吸附在吸附剂表面,从而实现吸附脱硫效果。此类脱硫吸附剂的主要吸附组分为活性金属离子修饰的分子筛或活性炭材料等,具有吸附温度低、无副反应、易于再生等优点,但其吸附原理也导致了吸附容量低、产品收率低、脱硫效果较差等缺点,因此限制了其在实际工业生产中的应用。而催化反应吸附脱硫过程是通过催化反应仅将含硫化合物分子中的硫原子转移至吸附剂当中。催化反应脱硫吸附剂也称作吸附脱硫催化剂,其主要吸附组分为过渡金属氧化物。由于吸附脱硫催化剂中吸附组分的体相部分也能参与反应,因此具有较高的吸附容量,并且化学反应的推动力也使其具有很好的脱硫效果。
目前工业上所使用吸附脱硫催化剂大部分以zno作为主要吸附组分、以ni组分等作为促进助剂,具有价格低廉、易于成型等优点。此类吸附脱硫催化剂的制备方法已有不少专利公开:cn101249440b、cn105728027a分别公开了通过共沉淀方法制备的以nio、zno为主要组分的吸附脱硫催化剂;cn107159096a、cn101619231b分别公开了通过浸渍法将ni组分负载至含zno载体上制备的脱硫吸附剂;cn107159096a、cn101619231b分别公开了通过浸渍法将ni组分负载至含zno载体上制备的脱硫吸附剂;cn104056632b公开了通过将含ni活性组分采用混捏法或浸渍法负载到含zn硫吸附剂上制备的吸附脱硫催化剂;cn102430412b公开了通过水热处理或浸渍法制备的含ni、zn等活性组分的汽油高选择性吸附脱硫剂;cn1326977c、cn101940908b、cn101905161b分别公开了通过将nio、zno等组分混合成型制备的脱硫吸附剂。除此之外,还有专利公开了向吸附脱硫催化剂中引入分子筛或碳基大孔材料来调节孔道结构,对其吸附脱硫性能也有一定的促进效果:cn102343276b、cn103657709a、cn103623773b、cn103642520b分别公开了使用zsm-5分子筛和zno等混合成型制备的脱硫吸附剂;cn105617984a、cn105688801a、cn105694948b、cn105694949b分别公开了使用sba-15分子筛和zno、nio等组分混合成型制备的脱硫吸附剂;cn102463098b、cn102463099b、cn102463100b分别公开了使用ael结构的磷铝酸盐分子筛和氧化锌、还原态促进剂等混合成型制备的脱硫吸附剂;cn104028215b、cn104028216b、cn104028217b分别公开了使用炭气凝胶或活性炭和氧化锌、氧化镍等混合成型制备的脱硫吸附剂。除此之外,us6346190公开了由美国康菲公司开发的s-zorb吸附脱硫技术,其中吸附剂由zno、nio和al2o3组成,采用流化床反应器和连续再生技术,该技术辛烷值损失小、氢耗低、脱硫效果好,但是投资成本高,不适合小型炼油企业。以上专利公开的吸附脱硫催化剂都以氧化锌作为主要吸附组分、以氧化镍等促进助剂提高其脱硫催化活性,同时可选地加入一些其他结构助剂等调节改善吸附脱硫催化剂的结构性质和催化性能。
虽然通过以上方法制备得到的以zn和ni等氧化物为主要组分的成型催化剂用于处理含硫原料能够实现一定的脱硫效果,然而在实际应用过程中存在如下问题:
1、吸附组分zno和载体al2o3之间存在较强的相互作用,在吸附脱硫和反复再生过程中的高温条件下易形成znal2o4尖晶石相,使得部分zno失去作用,导致吸附脱硫催化剂硫容不断下降(参考cn104028215b、cn104028216b、cn104028217b);
2、活性组分nio和载体al2o3之间也存在较强的相互作用,并且在吸附脱硫和反复再生过程中的高温条件下易形成nial2o4尖晶石相,使得ni物种难以被还原,导致吸附脱硫催化剂活性不断下降(参考cn104028215b、cn104028216b、cn104028217b);
3、吸附组分zno和活性组分nio也存在一定的相互作用,在催化剂预处理阶段也会有少部分还原生成nizn合金相,而过多的nizn合金相使得催化剂表面的活性ni物种发生聚集,也会导致催化剂的吸附脱硫性能下降(参考《石油学报(石油加工)》,2015年6月,第31卷,第3期)。
前期工作已经在专利中公开了不同组成的吸附脱硫催化剂及其制备方法(cn101450302b、cn103721668b、cn104707565a),在较为温和的条件下可实现柴油的超深度加氢脱硫,但根据实际生产应用和后续对催化剂的表征结果,发现了催化剂在使用和再生过程中由于活性组分分散度下降和组分之间相互作用导致脱硫性能有所下降。
综上所述,对于以zn和ni等氧化物为主要组分、以al2o3为载体的吸附脱硫催化剂,若要提升其脱硫活性和硫容等性能,以及避免催化剂在再生过程中的性能损失,除了通过引入结构助剂等改善颗粒分散度和孔道结构性质,有利于原料的扩散,还需要提升催化剂表面ni活性相的分散度,增加活性位数目,并且还要抑制al2o3载体、zn和ni组分之间的相互作用,防止由于生成尖晶石相和合金相而产生的活性组分损失和聚集等不利影响。本专利所公开的吸附脱硫催化剂是在前期工作的基础上加以改进,借鉴了之前吸附脱硫催化剂的基本合成方法,同时通过特殊的方法引入新的促进组分,同时提高了活性组分的分散度并抑制了组分之间的相互作用,合成了具有更优性能的吸附脱硫催化剂。
技术实现要素:
本发明的目的在于提供一种活性组分高分散的吸附脱硫催化剂。
本发明的另一目的在于提供一种所述催化剂的制备方法。
本发明的又一目的在于提供所述催化剂在含硫气体或液体超深度吸附脱硫中的应用。
本发明所述催化剂的技术特征包含以下几个方面:
所述吸附脱硫催化剂由至少一种+2价金属氧化物、一种+3价金属氧化物、一种iia族金属氧化物、至少一种ivb族金属氧化物和至少一种viii族金属氧化物组成。
其中:
所述至少一种+2价金属选自zn、cu、mn中的一种或二种以上;
所述至少一种viii族金属选自ni、co中的一种或二种;
所述至少一种ivb族金属选自ti、zr中的一种或二种;
所述+3价金属选自al;
所述iia族金属选自mg。
所述吸附脱硫催化剂的制备方法如下:
a)将至少一种+2价金属的可溶性盐溶液与沉淀剂溶液混合进行沉淀反应,老化并将得到的沉淀过滤、洗涤;
b)将步骤a得到的沉淀加入至少一种ivb族金属的可溶性盐溶液中进行处理,使得ivb族金属组分在步骤a得到的沉淀颗粒表面形成覆盖层,然后过滤并将得到的固体洗涤、干燥;
c)将步骤b得到的固体、一种iia族金属的可溶性盐溶液、含有+3价金属的粘合剂和造孔剂混合均匀后挤条成型,经干燥、焙烧后得到成型催化剂前体;
d)使用至少一种viii族金属的可溶性盐溶液对步骤c得到的成型催化剂前体进行浸渍,经干燥、焙烧后得到活性组分高分散的吸附脱硫催化剂。
以金属氧化物计,所述催化剂中含有30-80wt.%的+2价金属、1-30wt.%的viii族金属、0.5-5wt.%的ivb族金属、5-30wt.%的+3价金属和1-10wt.%的iia族金属。
所述吸附脱硫催化剂的比表面积为30-100m2/g,孔容为0.2-0.8ml/g。
所述催化剂的制备方法中:
步骤a中所述至少一种+2价金属的可溶性盐为硝酸锌、氯化锌、醋酸锌、硫酸锌、硝酸铜、氯化铜、醋酸铜、硫酸铜、醋酸锰、氯化锰、硝酸锰、硫酸锰中的一种或二种以上的组合;
步骤a中所述沉淀剂为为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、氨水、尿素、碳酸铵中的一种或二种以上的组合;
步骤b中所述至少一种ivb族金属的可溶性盐为硝酸钛、硫酸钛、硫酸氧钛、四氯化钛、硝酸锆、醋酸锆、硫酸锆中的一种或二种以上的组合;
步骤c中所述的一种iia族金属的可溶性盐为硝酸镁、硫酸镁、氯化镁中的一种或二种以上的组合;
步骤c中所述含有+3价金属的粘合剂为无定型氧化铝、拟薄水铝石、高岭土、硅藻土、蒙脱土中的一种或二种以上的组合;
步骤c中所述造孔剂为甲基纤维素、柠檬酸、石墨粉、栲胶中的一种或二种以上的组合;
步骤d中所述至少一种viii族金属的可溶性盐为硝酸镍、醋酸镍、硫酸镍、氯化镍、硝酸钴、醋酸钴、硫酸钴、氯化钴中的一种或二种以上的组合。
所述催化剂的制备方法中:
步骤a中所述至少一种+2价金属的可溶性盐溶液的浓度为0.1-2.0mol/l;
优选地,步骤a中所述至少一种+2价金属的可溶性盐溶液的浓度为0.5-1.0mol/l;
步骤a中所述沉淀剂溶液的浓度为0.1-2.0mol/l;
优选地,步骤a中所述沉淀剂溶液的浓度为0.5-1.0mol/l;
步骤b中所述至少一种ivb族金属的可溶性盐溶液的浓度为0.001-0.100mol/l;
优选地,步骤b中所述至少一种ivb族金属的可溶性盐溶液的浓度为0.005-0.020mol/l;
步骤c中所述一种iia族金属的可溶性盐溶液的浓度为0.1-5.0mol/l;
优选地,步骤c中所述一种iia族金属的可溶性盐溶液的浓度为0.5-2.0mol/l;
步骤d中所述至少一种viii族金属的可溶性盐溶液的浓度为0.1-10.0mol/l;
优选地,步骤d中所述至少一种viii族金属的可溶性盐溶液的浓度为5.0-10.0mol/l。
所述催化剂的制备方法中:
步骤a中所述沉淀反应和老化温度为40-100℃,老化时间为2-10h,老化过程反应体系ph为6-10;
优选地,步骤a中所述沉淀反应和老化温度为80-100℃,老化时间为2-4h,老化过程反应体系ph为8-10;
步骤b中所述处理温度为80-160℃,处理时间为5-40h,干燥温度为80-120℃,干燥时间为2-6h;
优选地,步骤b中所述处理温度为100-120℃,处理时间为6-20h,干燥温度为100-120℃,干燥时间为4-6h;
步骤c中所述干燥温度为80-120℃,干燥时间为2-6小时;焙烧温度为300-500℃,焙烧时间为4-10h;
优选地,步骤c中所述干燥温度为100-120℃,干燥时间为4-6小时;焙烧温度为350-400℃,焙烧时间为4-6h;
步骤d中所述干燥温度为80-120℃,干燥时间为2-6小时;焙烧温度为300-500℃,焙烧时间为4-10h;
优选地,步骤d中所述干燥温度为100-120℃,干燥时间为4-6小时;焙烧温度为350-400℃,焙烧时间为4-6h;
所述吸附脱硫催化剂用于含硫气体或液体的吸附脱硫过程,其中:
所述含硫气体为含硫的n2、ar、h2、co、co2、甲烷、乙烷、乙烯、丙烷、丙烯、丁烷、正丁烯、异丁烯中的一种或二种以上的组合;
所述含硫液体为含硫的异戊烷、异戊烯、环戊烷、环戊烯、甲基环戊烷、甲基环戊烯、正己烷、正己烯、环己烷、环己烯、甲基环己烷、甲基环己烯、正庚烷、正庚烯、汽油、柴油中的一种或二种以上的组合。
所述催化剂在对含硫气体或液体进行吸附脱硫前需按照如下条件进行预处理:
气氛为h2或n2和h2的混合气,其中h2分压为0.01-0.50mpa,预处理温度为300-450℃,h2的体积空速为100-1000h-1,预处理时间为2-36h;
优选地,h2分压为0.05-0.20mpa,预处理温度为400-450℃,h2的体积空速为300-500h-1,预处理时间为20-30h。
所述催化剂在对含硫液体进行吸附脱硫时的反应条件如下:
气氛为h2或n2和h2的混合气,其中h2分压为0.05-2.0mpa,反应温度为300-450℃,h2与含硫液体的体积比为500:1-10:1v/v,含硫液体的体积空速为1.0-10.0h-1;
优选地,h2分压为0.05-1.0mpa,反应温度为300-400℃,h2与含硫液体的体积比为100:1-10:1v/v,含硫液体的体积空速为1.0-5.0h-1。
所述催化剂在对含硫气体进行吸附脱硫时的反应条件如下:
气氛为含硫气体,压力为0.05-2.0mpa,反应温度为200-450℃,含硫气体的体积空速为50.0-5000.0h-1;
优选地,压力为0.30-2.0mpa,反应温度为250-350℃,含硫气体的体积空速为200.0-2500.0h-1。
当吸附脱硫反应进行一段时间后,吸附脱硫催化剂会逐渐失活,导致吸附脱硫反应无法继续进行,此时应当对失活催化剂进行再生处理,使其恢复吸附脱硫活性,具体再生处理条件如下:
气氛为n2和o2的混合气,体系压力为0.1-1.0mpa,其中o2的体积百分比为0.1-10.0%,气体的体积空速为100-6000h-1,再生温度为240-600℃,再生时间为12-72h;
优选地,体系压力为0.1-0.5mpa,其中o2的体积百分比为1.0-3.0%,气体的体积空速为300-3000h-1,再生温度为400-550℃,再生时间为20-40h。
所述催化剂具有较高的吸附脱硫活性和硫容,其相对脱硫活性不低于99.5%,硫容不低于16.0%,并且再生后催化剂的相对脱硫活性恢复至不低于99.0%,硫容恢复至不低于15.0%。
所述催化剂的相对脱硫活性按以下公式计算:
其中:
sf是含硫气体或液体的硫含量,单位是mg/l;
sp是使用催化剂吸附脱硫后气体或液体产品的硫含量,单位是mg/l。
所述催化剂的硫容按以下公式计算:
其中:
lhsv是气体或液体的体积空速,单位是h-1;
t是吸附脱硫过程从开始至气体或液体产品硫含量高于10mg/l为止所经历的时间,单位是h;
sf是含硫气体或液体的硫含量,单位是mg/l;
sp是使用催化剂吸附脱硫后气体或液体产品产品的硫含量,单位是mg/l;
ρcat是吸附脱硫催化剂的堆积密度,单位是kg/l。
本发明所提供的吸附脱硫催化剂与现有公知技术相比,具有如下优点:
1)通过特殊的合成方法引入助剂组分,使得助剂组分在+2价金属氧化物颗粒表面形成覆盖层,通过助剂组分和viii族金属氧化物之间的相互作用提高活性组分在颗粒表面的分散度,从而形成更多的催化反应活性中心,提高催化剂的吸附脱硫活性;
2)由于颗粒表面的助剂组分覆盖层分别与al2o3载体和viii族金属氧化物之间存在相互作用,能够在一定程度上削弱al2o3载体和viii族金属氧化物之间的相互作用,避免由于在吸附脱硫和再生过程中两者反应生成尖晶石相而导致的催化剂活性降低;
3)通过所述合成方法在+2价金属氧化物颗粒表面形成的助剂组分覆盖层减少了+2价金属氧化物与al2o3载体的接触机会,能够有效避免由于在吸附脱硫和再生过程中两者反应生成尖晶石相而导致的催化剂活性和硫容降低;
4)通过所述合成方法在+2价金属氧化物颗粒表面形成的助剂组分覆盖层还减少了+2价金属氧化物与viii族金属氧化物的接触机会,能够有效避免由于在预处理过程中两者反应生成合金相而导致的催化剂活性和硫容降低。
附图说明
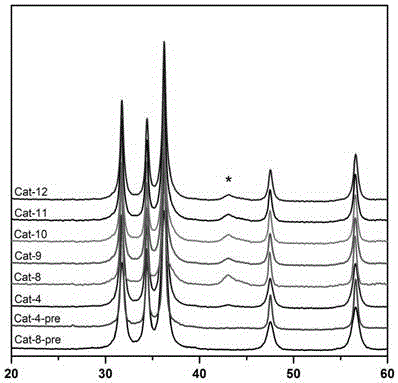
图1是实施例4和8中成型催化剂前体以及实施例4、8、9、10、11和12中成型吸附脱硫催化剂的xrd。此表征结果反映了对应样品中所含的物相及其结晶度,通过对比可以比较不同样品所含物相的分散度。其中:
cat-4-pre是实施例4中的成型催化剂前体;
cat-8-pre是实施例8中的成型催化剂前体;
cat-4是实施例4中的成型吸附脱硫催化剂;
cat-8是实施例8中的成型吸附脱硫催化剂;
cat-9是实施例9中的成型吸附脱硫催化剂;
cat-10是实施例10中的成型吸附脱硫催化剂;
cat-11是实施例11中的成型吸附脱硫催化剂;
cat-12是实施例12中的成型吸附脱硫催化剂。
图1中位于2θ=31.8°、34.4°、36.3°、47.5°和56.6°的衍射峰属于zno物相,位于2θ=43.1°的衍射峰(图中*标记处)属于nio物相。其中所有样品中属于zno的衍射峰无明显差异,说明以上实施例中不同的合成方法和viii族金属组分的浸渍过程对zno晶体颗粒的结构和分散度基本无影响。通过对比cat-4、cat-8、cat-9、cat-10、cat-11、cat-12中2θ=43.1°属于nio衍射峰的差异,可以发现:
1)当未向催化剂中引入ivb族金属组分作为促进助剂时(对应cat-8),nio活性组分的分散度最差;
2)当向催化剂中引入ivb族金属组分作为促进助剂,但其引入方法不同于本发明所述的合成方法时(对应cat-9、cat-10、cat-11、cat-12),nio活性组分的分散度有所提升,但效果有限;
3)当向催化剂中引入ivb族金属组分作为促进助剂,并且引入方法与本发明所述的合成方法相同时(对应cat-4),nio活性组分的分散度有非常大的提升,效果在所有实施例中最为明显。
图2是实施例4、8、9、10、11和12中成型吸附脱硫催化剂的tpr。此表征结果反映了对应样品中活性组分nio的还原温度,通过对比可以比较不同样品中nio与其他组份之间的相互作用。图中样品的命名与图1相同。从该结果可以看出nio的还原过程主要发生在两个温度区域:即350℃-500℃的低温区和550℃以上的高温区。由于当nio与al2o3载体接触时会产生较强的相互作用,导致nio不易被还原,因此可以认为低温区的还原峰对应未与al2o3载体接触作用的nio的还原过程,而高温区的还原峰对应与al2o3载体接触作用的nio的还原过程,并且两个还原峰的面积代表了对应nio的含量。通过对比不同样品的结果可以发现:
1)当未向催化剂中引入ivb族金属组分作为促进助剂时(对应cat-8),与al2o3载体接触作用的nio活性组分含量最高;
2)当向催化剂中引入ivb族金属组分作为促进助剂,但其引入方法不同于本发明所述的合成方法时(对应cat-9、cat-10、cat-11、cat-12),与al2o3载体接触作用的nio活性组分含量有所下降,未与al2o3载体接触作用的nio活性组分含量有所提升,但效果有限;
3)当向催化剂中引入ivb族金属组分作为促进助剂,并且引入方法与本发明所述的合成方法相同时(对应cat-4),与al2o3载体接触作用的nio活性组分含量明显下降,未与al2o3载体接触作用的nio活性组分含量明显提升,效果在所有实施例中最为明显。这里由于nio与ivb族金属组分之间也存在较弱的相互作用,导致低温区的还原峰温度略微有所提升。
图3是实施例4、8、9、10、11和12中成型吸附脱硫催化剂的tps。此表征结果反映了对应样品中吸附组分zno的硫化温度,通过对比可以比较不同样品中zno与其他组份之间的相互作用。图中样品的命名与图1相同。从该结果可以看出zno的硫化过程主要发生在两个温度区域:即100℃-200℃的低温区和200℃-300℃的高温区。由于当zno与al2o3载体接触时会产生较强的相互作用,导致zno不易被硫化,因此可以认为低温区的还原峰对应未与al2o3载体接触作用的zno的硫化过程,而高温区的还原峰对应与al2o3载体接触作用的zno的硫化过程,并且两个还原峰的面积代表了对应zno的含量。通过对比不同样品的结果可以发现:
1)当未向催化剂中引入ivb族金属组分作为促进助剂时(对应cat-8),与al2o3载体接触作用的zno吸附组分含量最高;
2)当向催化剂中引入ivb族金属组分作为促进助剂,但其引入方法不同于本发明所述的合成方法时(对应cat-9、cat-10、cat-11、cat-12),与al2o3载体接触作用的zno吸附组分含量有所下降,未与al2o3载体接触作用的zno吸附组分含量有所提升,但效果有限;
3)当向催化剂中引入ivb族金属组分作为促进助剂,并且引入方法与本发明所述的合成方法相同时(对应cat-4),与al2o3载体接触作用的zno吸附组分含量明显下降,未与al2o3载体接触作用的zno吸附组分含量明显提升,效果在所有实施例中最为明显。
综上所述,当采用本发明所述的合成方法向催化剂中引入ivb族金属组分作为促进助剂时,1)能够有效提升nio活性组分的分散度,有助于形成更多的催化反应活性中心并提高催化剂的吸附脱硫活性;2)能够有效抑制nio活性组分与al2o3载体之间的相互作用,使得nio活性组分在预处理过程中更易被还原活化,产生催化活性中心;3)能够有效抑制zno吸附组分与al2o3载体之间的相互作用,使得zno吸附组分在吸附脱硫过程中更易与含硫化合物反应,起到脱硫效果。
具体实施方式
为了进一步说明本发明,列举以下实施例,但并不限制权利要求所定义的发明范围。
本发明根据实验结果提出一种由至少一种+2价金属、一种+3价金属、一种iia族金属、至少一种ivb族金属和至少一种viii族金属组成的混合金属氧化物吸附脱硫催化剂及其制备方法,其中所述+2价金属选自zn、cu、mn,viii族金属选自ni、co,ivb族金属选自ti、zr,+3价金属选自al,iia族金属选自mg。在这里为了简明起见,列举了部分金属选取的实施例,但并不意味其余的金属选取不能实现本发明。
实施例1
本实施例说明使用本发明所述合成方法制备金属组分为ni、ti、zn、al、mg的吸附脱硫催化剂:
a)称取58.0g六水合硝酸锌,加入300ml去离子水配成溶液,加热至80℃并恒温,然后称取30.0g碳酸钠,加入300ml去离子水配成溶液,再将碳酸钠溶液在搅拌下加入至硝酸锌溶液中至ph为10,此时生成白色沉淀,最后在80℃恒温下搅拌老化2h后将沉淀过滤并用去离子水洗涤3遍;
b)称取0.6g硫酸氧钛,加入300ml去离子水配成溶液,然后加入步骤a得到的白色沉淀,于水热釜中120℃处理10h,再将沉淀过滤并用去离子水洗涤3遍,最后在110℃干燥6h;
c)称取3.8g六水合硝酸镁,加入8ml去离子水配成溶液,然后加入步骤b得到的沉淀、8.5g拟薄水铝石和0.1g甲基纤维素混合均匀并挤条成型,再在110℃干燥6h,最后在350℃焙烧4h后得到成型催化剂前体;
d)称取23.4g六水合硝酸镍,加入10ml去离子水配成溶液,然后将硝酸镍溶液对步骤c得到的成型催化剂前体进行浸渍,再在110℃干燥6h,最后在350℃焙烧4h后得到成型吸附脱硫催化剂,记为cat-1。经低温氮吸附测定,催化剂的比表面积为52m2/g,孔容为0.42ml/g。
实施例2
本实施例说明使用本发明所述合成方法制备金属组分为ni、ti、mn、al、mg的吸附脱硫催化剂:
除了使用44.3g四水合氯化锰代替实施例1中使用的58.0g六水合硝酸锌,以与实施例1中所述的相同方式制备得到成型吸附脱硫催化剂,记为cat-2。经低温氮吸附测定,催化剂的比表面积为67m2/g,孔容为0.55ml/g。
实施例3
本实施例说明使用本发明所述合成方法制备金属组分为ni、zr、mn、al、mg的吸附脱硫催化剂:
除了使用44.3g四水合氯化锰代替实施例1中使用的58.0g六水合硝酸锌、使用1.0g五水合硝酸锆代替实施例1中使用的0.6g硫酸氧钛,以与实施例1中所述的相同方式制备得到成型吸附脱硫催化剂,记为cat-3。经低温氮吸附测定,催化剂的比表面积为71m2/g,孔容为0.56ml/g。
实施例4
本实施例说明使用本发明所述合成方法制备金属组分为ni、zr、zn、al、mg的吸附脱硫催化剂:
除了使用1.0g五水合硝酸锆代替实施例1中使用的0.6g硫酸氧钛,以与实施例1中所述的相同方式制备得到成型吸附脱硫催化剂,记为cat-4。经低温氮吸附测定,催化剂的比表面积为56m2/g,孔容为0.43ml/g。
实施例5
本实施例说明使用本发明所述合成方法制备金属组分为ni、zr、zn、al、mg的吸附脱硫催化剂:
除了使用1.5g五水合硝酸锆代替实施例1中使用的0.6g硫酸氧钛,以与实施例1中所述的相同方式制备得到成型吸附脱硫催化剂,记为cat-5。经低温氮吸附测定,催化剂的比表面积为58m2/g,孔容为0.44ml/g。
实施例6
本实施例说明使用本发明所述合成方法制备金属组分为ni、zr、zn、al、mg的吸附脱硫催化剂:
除了使用2.0g五水合硝酸锆代替实施例1中使用的0.6g硫酸氧钛,以与实施例1中所述的相同方式制备得到成型吸附脱硫催化剂,记为cat-6。经低温氮吸附测定,催化剂的比表面积为51m2/g,孔容为0.42ml/g。
实施例7
本实施例说明使用本发明所述合成方法制备金属组分为ni、zr、zn、al、mg的吸附脱硫催化剂:
除了使用0.5g五水合硝酸锆代替实施例1中使用的0.6g硫酸氧钛,以与实施例1中所述的相同方式制备得到成型吸附脱硫催化剂,记为cat-7。经低温氮吸附测定,催化剂的比表面积为56m2/g,孔容为0.44ml/g。
实施例8
本实施例说明不含ivb族金属组分作为促进助剂的吸附脱硫催化剂,其合成方法省去了本发明所述合成方法中的步骤b,以作对比:
a)称取58.0g六水合硝酸锌,加入300ml去离子水配成溶液,加热至80℃并恒温,然后称取30.0g碳酸钠,加入300ml去离子水配成溶液,再将碳酸钠溶液在搅拌下加入至硝酸锌溶液中至ph为10,此时生成白色沉淀,最后在80℃恒温下搅拌老化2h,将沉淀过滤后用去离子水洗涤3遍并在110℃干燥6h;
b)称取3.8g六水合硝酸镁,加入8ml去离子水配成溶液,然后加入步骤a得到的沉淀、8.5g拟薄水铝石和0.1g甲基纤维素混合均匀并挤条成型,再在110℃干燥6h,最后在350℃焙烧4h后得到成型催化剂前体;
c)称取23.4g六水合硝酸镍,加入10ml去离子水配成溶液,然后将硝酸镍溶液对步骤b得到的成型催化剂前体进行浸渍,再在110℃干燥6h,最后在350℃焙烧4h后得到成型吸附脱硫催化剂,记为cat-8。经低温氮吸附测定,催化剂的比表面积为46m2/g,孔容为0.35ml/g。
实施例9
本实施例说明组成与实施例4相同的吸附脱硫催化剂,但其ivb族金属的引入方法与本发明所述方法不同,以作对比:
a)称取58.0g六水合硝酸锌和1.0g五水合硝酸锆,加入300ml去离子水配成溶液,加热至80℃并恒温,然后称取30.0g碳酸钠,加入300ml去离子水配成溶液,再将碳酸钠溶液在搅拌下加入至硝酸锌溶液中至ph为10,此时生成白色沉淀,最后在80℃恒温下搅拌老化2h,将沉淀过滤后用去离子水洗涤3遍并在110℃干燥6h;
b)称取3.8g六水合硝酸镁,加入8ml去离子水配成溶液,然后加入步骤a得到的沉淀、8.5g拟薄水铝石和0.1g甲基纤维素混合均匀并挤条成型,再在110℃干燥6h,最后在350℃焙烧4h后得到成型催化剂前体;
c)称取23.4g六水合硝酸镍,加入10ml去离子水配成溶液,然后将硝酸镍溶液对步骤b得到的成型催化剂前体进行浸渍,再在110℃干燥6h,最后在350℃焙烧4h后得到成型吸附脱硫催化剂,记为cat-9。经低温氮吸附测定,催化剂的比表面积为60m2/g,孔容为0.46ml/g。
实施例10
本实施例说明组成与实施例4相同的吸附脱硫催化剂,但其ivb族金属的引入方法与本发明所述方法不同,以作对比:
a)称取58.0g六水合硝酸锌,加入300ml去离子水配成溶液,加热至80℃并恒温,然后称取30.0g碳酸钠,加入300ml去离子水配成溶液,再将碳酸钠溶液在搅拌下加入至硝酸锌溶液中至ph为10,此时生成白色沉淀,最后在80℃恒温下搅拌老化2h,将沉淀过滤后用去离子水洗涤3遍并在110℃干燥6h;
b)称取3.8g六水合硝酸镁和1.0g五水合硝酸锆,加入8ml去离子水配成溶液,然后加入步骤a得到的沉淀、8.5g拟薄水铝石和0.1g甲基纤维素混合均匀并挤条成型,再在110℃干燥6h,最后在350℃焙烧4h后得到成型催化剂前体;
c)称取23.4g六水合硝酸镍,加入10ml去离子水配成溶液,然后将硝酸镍溶液对步骤b得到的成型催化剂前体进行浸渍,再在110℃干燥6h,最后在350℃焙烧4h后得到成型吸附脱硫催化剂,记为cat-10。经低温氮吸附测定,催化剂的比表面积为49m2/g,孔容为0.38ml/g。
实施例11
本实施例说明组成与实施例4相同的吸附脱硫催化剂,但其ivb族金属的引入方法与本发明所述方法不同,以作对比:
a)称取58.0g六水合硝酸锌,加入300ml去离子水配成溶液,加热至80℃并恒温,然后称取30.0g碳酸钠,加入300ml去离子水配成溶液,再将碳酸钠溶液在搅拌下加入至硝酸锌溶液中至ph为10,此时生成白色沉淀,最后在80℃恒温下搅拌老化2h,将沉淀过滤后用去离子水洗涤3遍并在110℃干燥6h;
b)称取3.8g六水合硝酸镁,加入8ml去离子水配成溶液,然后加入步骤a得到的沉淀、8.5g拟薄水铝石和0.1g甲基纤维素混合均匀并挤条成型,再在110℃干燥6h,最后在350℃焙烧4h后得到成型催化剂前体;
c)称取23.4g六水合硝酸镍和1.0g五水合硝酸锆,加入10ml去离子水配成溶液,然后将硝酸镍溶液对步骤b得到的成型催化剂前体进行浸渍,再在110℃干燥6h,最后在350℃焙烧4h后得到成型吸附脱硫催化剂,记为cat-11。经低温氮吸附测定,催化剂的比表面积为44m2/g,孔容为0.34ml/g。
实施例12
本实施例说明组成与实施例4相同的吸附脱硫催化剂,但其ivb族金属的引入方法与本发明所述方法不同,以作对比:
a)称取58.0g六水合硝酸锌、23.4g六水合硝酸镍、加入300ml去离子水配成溶液、1.0g五水合硝酸锆,加热至80℃并恒温,然后称取30.0g碳酸钠,加入300ml去离子水配成溶液,再将碳酸钠溶液在搅拌下加入至硝酸锌溶液中至ph为10,此时生成白色沉淀,最后在80℃恒温下搅拌老化2h,将沉淀过滤后用去离子水洗涤3遍并在110℃干燥6h;
b)称取3.8g六水合硝酸镁,加入8ml去离子水配成溶液,然后加入步骤a得到的沉淀、8.5g拟薄水铝石和0.1g甲基纤维素混合均匀并挤条成型,再在110℃干燥6h,最后在350℃焙烧4h后得到成型吸附脱硫催化剂,记为cat-12。经低温氮吸附测定,催化剂的比表面积为65m2/g,孔容为0.50ml/g。
实施例13
本实施例说明新鲜催化剂的预处理过程以及对汽油吸附脱硫活性和硫容的评价:
a)分别称取2.0g实施例1-12中制备的成型吸附脱硫催化剂cat-1至cat-12装填在固定床反应器中,使用h2气氛在温度为400℃、h2分压为0.1mpa、h2体积空速为360h-1的条件下对其进行预处理,处理时间为24h;
b)使用h2气氛在温度为350℃、h2分压为1.0mpa、原料的体积空速为2.0h-1、h2与原料的体积比为25:1v/v的条件下通入硫含量为200mg/l的汽油原料进行吸附脱硫,根据反应进行24h时汽油产品的硫含量计算吸附脱硫催化剂的相对活性,结果如表1所示;
c)当汽油产品硫含量高于10mg/l时停止吸附脱硫反应,根据反应维持的时间计算吸附脱硫催化剂的硫容,结果如表1所示。
实施例14
本实施例说明失活催化剂的再生处理过程以及再生催化剂对汽油吸附脱硫的活性和硫容评价:
a)使用o2体积百分比为2.0%的n2和o2的混合气在体系压力为0.1mpa、气体的体积空速为1200h-1、温度为460℃的条件下,分别对实施例13中失活的吸附脱硫催化剂cat-1至cat-12进行再生处理,再生时间为36h;
b)使用和实施例13中相同的预处理方法对再生催化剂进行预处理,并使用和实施例13中相同的评价方法评价再生催化剂的吸附脱硫活性和硫容,结果如表2所示。
实施例15
本实施例说明新鲜催化剂cat-4对含硫n2吸附脱硫活性和硫容的评价:
a)称取1.0g实施例4中制备的成型吸附脱硫催化剂cat-4装填在固定床反应器中,使用和实施例13中相同的预处理方法进行预处理;
b)在温度为300℃、压力为0.5mpa、原料的体积空速为1200.0h-1的条件下通入含有1.0vol.%h2s的n2进行吸附脱硫,根据反应进行24h时气体产品的硫含量计算吸附脱硫催化剂的相对活性,结果如表3所示;
c)当气体产品硫含量高于0.01vol.%时停止吸附脱硫反应,根据反应维持的时间计算脱硫吸附剂的硫容,结果如表3所示。
实施例16
本实施例说明再生催化剂cat-4对含硫n2吸附脱硫活性和硫容的评价:
a)使用和实施例14中相同的再生方法对实施例15中失活的吸附脱硫催化剂cat-4进行再生处理;
b)使用和实施例13中相同的预处理方法对再生催化剂进行预处理,并使用和实施例15中相同的评价方法评价再生催化剂的吸附脱硫活性和硫容,结果如表3所示。
实施例17
本实施例说明新鲜催化剂cat-4对含硫ar吸附脱硫活性和硫容的评价:
除了使用含有1.0vol.%h2s的ar代替实施例15中使用的含有1.0vol.%h2s的n2,以与实施例15中所述的评价方法评价催化剂的吸附脱硫活性和硫容,结果如表3所示。
实施例18
本实施例说明再生催化剂cat-4对含硫ar吸附脱硫活性和硫容的评价:
a)使用和实施例14中相同的再生方法对实施例17中失活的吸附脱硫催化剂cat-4进行再生处理;
b)使用和实施例13中相同的预处理方法对再生催化剂进行预处理,并使用和实施例17中相同的评价方法评价再生催化剂的吸附脱硫活性和硫容,结果如表3所示。
实施例19
本实施例说明新鲜催化剂cat-4对含硫h2吸附脱硫活性和硫容的评价:
除了使用含有1.0vol.%h2s的h2代替实施例15中使用的含有1.0vol.%h2s的n2,以与实施例15中所述的评价方法评价催化剂的吸附脱硫活性和硫容,结果如表3所示。
实施例20
本实施例说明再生催化剂cat-4对含硫h2吸附脱硫活性和硫容的评价:
a)使用和实施例14中相同的再生方法对实施例19中失活的吸附脱硫催化剂cat-4进行再生处理;
b)使用和实施例13中相同的预处理方法对再生催化剂进行预处理,并使用和实施例19中相同的评价方法评价再生催化剂的吸附脱硫活性和硫容,结果如表3所示。
表1、新鲜催化剂对汽油吸附脱硫的活性和硫容:
表2、再生催化剂对汽油吸附脱硫的活性和硫容:
表3、新鲜和再生cat-4对含硫气体吸附脱硫的活性和硫容:
- 还没有人留言评论。精彩留言会获得点赞!