一种凝胶制剂质量控制方法与流程
本发明涉及生物医药技术领域,具体涉及一种凝胶制剂质量控制方法。
背景技术:
凝胶制剂由药物溶解或均匀分散于凝胶中制成。因凝胶能与作用部位紧密黏附,有较好的生物相容性,多通过皮肤、黏膜给药,也可通过口服发挥药效。由于凝胶吸水溶胀后形成的水化凝胶层对药物有一定的控制释放作用,现广泛用于缓释、控释系统。对于制备得到的凝胶制剂,质量的控制十分重要,但是目前还没有一种有效的、针对主要成分是主要药物成分聚筛蕊提取物的凝胶制剂的质量控制方法。
技术实现要素:
针对现有技术的不足,本发明旨在提供一种凝胶制剂质量控制方法,以聚筛蕊药材、巴巴酸对照品检查制剂中主要药物成分聚筛蕊提取物,方法简便、宜操作,有较好的专属性。
为了实现上述目的,本发明采用如下技术方案:
一种凝胶制剂质量控制方法,包括如下步骤:
s1检查
1.1)对照品溶液制备
称取巴巴酸对照品加甲醇溶剂定容到容量瓶中,制备成每1ml含0.101mg的溶液,作为对照品溶液;
1.2)称取聚筛蕊药材置于锥形瓶中,加入丙酮溶液,置水浴锅加热回流,提取液滤过,水浴挥干,所得的浸膏用v甲醇:v丙酮=1:1的甲醇和丙酮的混合溶剂溶解,溶液滤过,取过滤液作为聚筛蕊对照药材溶液;
1.3)称取制备好的待测凝胶制剂置于三角烧瓶中,加入v乙腈:v0.1%磷酸=80:20的乙腈和0.1%磷酸的混合溶液,称重,加热超声20min,冷却,称定重量,用所述乙腈和0.1%磷酸的混合溶液补足损失的重量,离心12min,取上清液作为凝胶制剂供试品溶液;
1.4)按照薄层色谱的方法试验,将所述对照品溶液、聚筛蕊对照药材溶液和凝胶制剂供试品溶液,分别点于同一硅胶薄层板,用以v环己烷:v乙酸乙酯:v冰醋酸=4∶1∶0.25的环己烷、乙酸乙酯和冰醋酸混合的展开剂展开;取出晾干,喷以10%的硫酸溶液,加热显色;在薄层色谱展开相同的位置,对照品溶液、对照药材溶液和供试品溶液应显相同颜色斑点;
s2巴巴酸含量测定
2.1)确定色谱条件
diamonsilc18(2)柱:250mmx4.6mm,5μl;流动相:v乙腈:v0.1%磷酸=80:20的乙腈和0.1%磷酸的混合溶液;流速:1ml/min;检测波长:278nm;柱温:300℃;进样量:10μl;
2.2)对照品溶液的制备
称取巴巴酸对照品置于容量瓶中,加甲醇溶解并稀释至刻度,配成为0.1mg/ml的巴巴酸对照品溶液;
2.3)供试品的制备
称取待测凝胶制剂置于具塞锥形瓶中,加入所述乙腈和0.1%磷酸的混合溶液,并称定重量,超声20min,取出放冷,称定重量,以所述乙腈和0.1%磷酸的混合溶液补足重量,摇匀后,置入离心管中,高速离心,取上清液滤过,得供试品溶液;
2.4)按照高效液相色谱法,取步骤2.2)中得到的巴巴酸对照品溶液和步骤2.3)中得到的供试品溶液分别进样,按步骤2.1)中的色谱条件测定,测得的峰面积并计算含量;待测凝胶制剂所含浸提物以巴巴酸计算应不低于1.1mg/g。
进一步地,还包括有检查待测凝胶制剂形状的步骤:应呈淡绿色的粘稠体,稠度适中,涂展均匀。
进一步地,步骤1.1)具体如下:称取巴巴酸对照品1.01mg加甲醇溶剂定容到10ml的容量瓶中,制备成每1ml含0.101mg的溶液,作为对照品溶液。
进一步地,步骤1.2)具体为:称取10g的聚筛蕊药材置于250ml的锥形瓶中,加入150ml的丙酮溶液,置水浴锅加热回流2h,提取液滤过,水浴挥干,所得的浸膏以30ml所述甲醇和丙酮的混合溶剂溶解,溶液滤过,取过滤液作为聚筛蕊对照药材溶液。
进一步地,步骤1.3)具体为:称取制备好的待测凝胶制剂5.027g,置于三角烧瓶中,加入20ml的所述乙腈和0.1%磷酸的混合溶液,称重,加热超声20min,冷却,称定重量,用所述乙腈和0.1%磷酸的混合溶液补足损失的重量,离心12min,取上清液作为凝胶制剂供试品溶液。
进一步地,步骤2.2)具体为:精密称取巴巴酸对照品5.0mg,置于50ml的容量瓶中,加甲醇溶解并稀释至刻度,配成为0.1mg/ml的巴巴酸对照品溶液。
进一步地,步骤2.3)具体为:精密称取待测凝胶制剂1g,置于25ml的具塞锥形瓶中,精密加入20ml所述乙腈和0.1%磷酸的混合溶液,并称定重量,超声20min,取出放冷,称定重量,以所述乙腈和0.1%磷酸的混合溶液补足重量,摇匀后,置入50ml的离心管中,高速离心12min,取上清液微孔滤膜滤过,得供试品溶液。
本发明的有益效果在于:对于以聚筛蕊药材为主要成分的凝胶制剂给出了有效的质量控制方法,以聚筛蕊药材、巴巴酸对照品检查凝胶制剂中主要药物成分聚筛蕊提取物,方法简便、宜操作,有较好的专属性。
附图说明
图1为本发明的薄层色谱展开示意图;
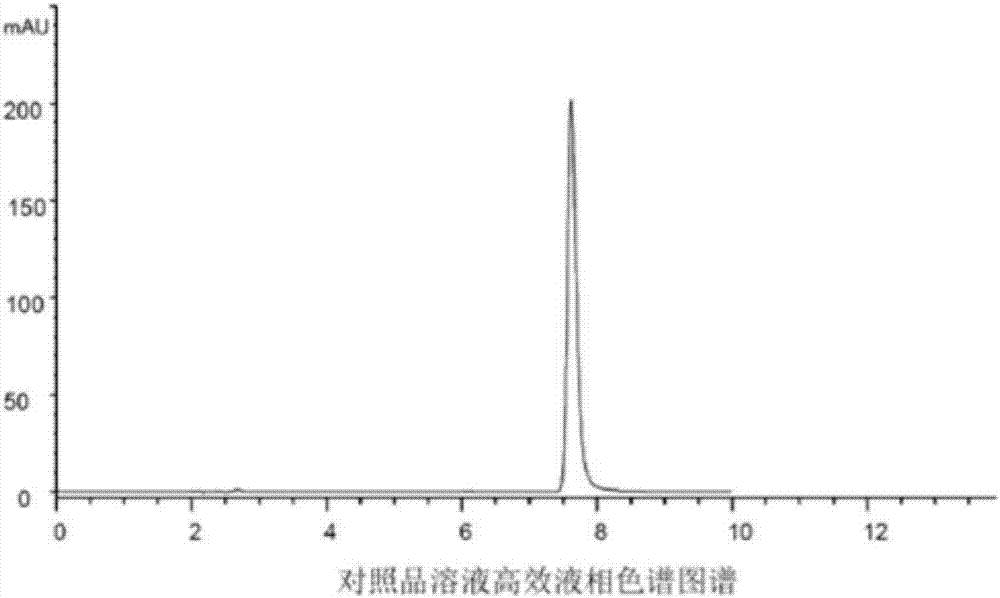
图2a、图2b和图2c分别是本发明的巴巴酸对照品溶液、凝胶制剂供试品溶液和阴性对照品溶液的高效液相色谱图谱。
图3为巴巴酸对照品溶液的紫外图谱;
图4a-图4e分别为不同浓度线性范围的考察的结果色谱图,图4f为线性关系考察曲线示意图;
图5a-图5f分别表示6次精密度测定的实验图谱;
图6a-6f分别表示在0h、2h、4h、6h、8h、10h将供试品溶液注入高效液相色谱仪的稳定性实验图谱;
图7a-图7f分别表示6次重复实验的实验图谱;
图8a-图8g分别为加样回收率实验的实验图谱,其中图8a所示为空白对照试验图谱,图8b-图8g分别表示6次实验的实验图谱。
图9为巴巴酸对照品制备流程图;
图10为巴巴酸对照品的质谱图。
具体实施方式
以下将结合附图对本发明作进一步的描述,需要说明的是,本实施例以本技术方案为前提,给出了详细的实施方式和具体的操作过程,但本发明的保护范围并不限于本实施例。
一种凝胶制剂质量控制方法,包括如下步骤:
s1检查
1.1)对照品和对照品溶液制备
巴巴酸对照品由贵州省中科院天然药物化学分析重点实验室分离纯化所得,纯度为97%,分离纯化流程图如图9所示,质谱图如图10所示。称取巴巴酸对照品加甲醇溶剂定容到容量瓶中,制备成每1ml含0.101mg的溶液,作为对照品溶液;
1.2)称取10g的聚筛蕊药材置于250ml的锥形瓶中,加入150ml的丙酮溶液,置水浴锅加热回流2h,提取液滤过,水浴挥干,所得的浸膏以30ml的甲醇∶丙酮=1∶1(体积比)的溶剂溶解,溶液滤过,取过滤液作为聚筛蕊对照药材溶液;
1.3)称取制备好的待测凝胶制剂5.027g,置于三角烧瓶中,加入20ml的乙腈∶0.1%磷酸=80∶20(体积比)的溶液,称重,加热超声20min,冷却,称定重量,用乙腈∶0.1%磷酸=80∶20(体积比)的溶液补足损失的重量,离心12min,取上清液作为凝胶制剂供试品溶液;
1.4)按照薄层色谱的方法试验,将所述对照品溶液、聚筛蕊对照药材溶液和凝胶制剂供试品溶液,分别点于同一硅胶薄层板,以环己烷∶乙酸乙酯∶冰醋酸=4∶1∶0.25(体积比)的展开剂展开;取出晾干,喷以10%的硫酸溶液,加热显色;在薄层色谱展开相同的位置,对照品溶液、对照药材溶液和供试品溶液应显相同颜色斑点;
s2巴巴酸含量测定
2.1)确定色谱条件
diamonsilc18(2)柱:250mmx4.6mm,5μl;流动相:乙腈:0.1%磷酸溶液=80∶20(体积比);流速:1ml/min;检测波长:278nm;柱温:300℃;进样量:10μl;
2.2)对照品溶液的制备
精密称取巴巴酸对照品5.0mg,置于50ml的容量瓶中,加甲醇溶解并稀释至刻度,配成为0.1mg/ml的巴巴酸对照品溶液。
2.3)供试品的制备
精密称取待测凝胶制剂1g,置于25ml的具塞锥形瓶中,精密加入20ml乙腈∶0.1%磷酸=80∶20(体积比)的溶液,并称定重量,超声20min,取出放冷,称定重量,以乙腈∶0.1%磷酸=80∶20(体积比)的溶液补足重量,摇匀后,置入离心管中,高速离心,取上清液微孔滤膜滤过,得供试品溶液;
2.4)按照高效液相色谱法,取步骤2.2)中得到的巴巴酸对照品溶液和步骤2.3)中得到的供试品溶液分别进样,按步骤2.1)中的色谱条件测定,测得的峰面积并计算含量;凝胶制剂所含浸提物以巴巴酸计算应不低于1.1mg/g。
需要说明的是,在检查步骤和巴巴酸含量测定步骤之前,先检查凝胶制剂形状的步骤:凝胶剂应呈淡绿色的粘稠体,稠度适中,涂展均匀。
本发明适用于以聚筛蕊药材为主要成分的凝胶制剂的质量控制。在本实施例中,参与性能实验的凝胶制剂的组分以重量份1000份计包括:聚筛蕊提取物100份;卡波姆11份;丁二醇200份;甘油160份;吐温10份;ve0.25份;三乙醇胺1.2份;玫瑰精油1.5份;蒸馏水余量。
制备方法包括如下步骤:
s1称取聚筛蕊药材粉碎,以丙酮回流提取2h,过滤,浓缩成稠膏状,得到聚筛蕊提取物;聚筛蕊药材和丙酮的料液比为1:15(g/ml);
s2取卡波姆置入适量蒸馏水中,静置过夜,使其充分溶胀,然后往其中加入甘油、吐温、丁二醇和ve并搅匀;加入聚筛蕊提取物,搅匀;再加入玫瑰精油和三乙醇胺,调节ph至6.0~7.0;加蒸馏水调整到1000份,搅拌均匀,分装即得。
以下将通过实验对本发明方法的性能进行分析
一、关于检查步骤的性能:
1)巴巴酸对照品溶液制备
取巴巴酸对照品1.01mg加甲醇溶剂定容到10ml的容量瓶,制备成每1ml含0.101mg的溶液,作为对照品溶液。
2)对照药材溶液制备
称取10g的聚筛蕊药材置于250ml的锥形瓶中,加入150ml的丙酮溶液,置水浴锅加热回流2h,提取液滤过,水浴挥干,所得的浸膏以30ml的甲醇∶丙酮=1∶1(体积比)溶剂溶解,溶液滤过,取过滤液作为聚筛蕊对照药材溶液。
3)供试品溶液制备
称取制备好的凝胶制剂5.027g,置于三角烧瓶中,加入20ml的乙腈∶0.1%磷酸=80∶20(体积比)的溶液,称重,加热超声20min,冷却,称定重量,用乙腈∶0.1%磷酸=80∶20(体积比)的溶液补足损失的重量,离心12min,取上清液作为凝胶制剂供试品溶液。
4)阴性对照溶液制备
称取不含聚筛蕊的凝胶制剂5.033g在三角瓶中,加入20ml的乙腈∶0.1%磷酸=80∶20(体积比)的溶液,称重,加热超声20min,冷却,称定重量,用乙腈∶0.1%磷酸=80∶20(体积比)的溶液补足重量,离心12min,取上清液作为阴性对照品溶液。
5)薄层色谱展开
按照薄层色谱的方法试验,将上述对照品溶液、对照药材溶液、阴性对照溶液和供试品溶液,分别点于同一硅胶薄层板,以环己烷∶乙酸乙酯∶冰醋酸=4∶1∶0.25(体积比)为展开剂展开。取出晾干,喷以10%的硫酸溶液,加热显色,结果显示对照品溶液、聚筛蕊对照药材溶液和凝胶制剂供试品溶液显相同颜色斑点,阴性对照溶液在相同展开位置无斑点,如图1所示。其中1对应对照品溶液,2对应聚筛蕊对照药材溶液;3和4对应凝胶制剂供试品溶液,5对应阴性对照品。
二、关于巴巴酸含量检测步骤的性能:
1、实验条件:
1)色谱条件
diamonsilc18(2)柱(250mmx4.6mm5μl);流动相:乙腈-0.1%磷酸溶液(80∶20体积比);流速:1ml/min;检测波长:278nm;柱温:300℃;进样量:10μl。
2)测试溶液制备
对照品溶液的制备
精密称取巴巴酸对照品5.0mg,置于50ml的容量瓶中,加甲醇溶解并稀释至刻度,配成为0.1mg/ml的巴巴酸对照品溶液。巴巴酸对照品溶液的紫外图谱如图3所示。
供试品溶液的制备
精密称取凝胶制剂1g,置于25ml的具塞锥形瓶中,精密加入20ml的乙腈∶0.1%磷酸=80∶20(体积比)的溶液,并称定重量,超声20min,取出放冷,称定重量,以乙腈∶0.1%磷酸=80∶20(体积比)的溶液补足重量,摇匀后,置入50ml的离心管中,高速离心12min,取上清液微孔滤膜滤过,得供试品溶液。
阴性对照品溶液的制备
精密称取空白凝胶制剂(不含聚筛蕊)1g,置于25ml的具塞锥形瓶中,精密加入20ml的乙腈∶0.1%磷酸=80∶20(体积比)的溶液,并称定重量,超声20min,取出放冷,称定重量,以乙腈∶0.1%磷酸=80∶20(体积比)的溶液补足重量,摇匀后,置入50ml的离心管中,高速离心12min,取上清液微孔滤膜滤过,得阴性对照品溶液。
2、色谱条件研究
按上述色谱条件,取巴巴酸对照品溶液、供试品溶液和阴性对照品溶液分别注入色谱仪中,实验结果显示,供试品溶液中的主要成分出峰的保留时间和巴巴酸对照品溶液出峰的保留时间是一致的,阴性对照品溶液在相同保留时间未见吸收,如图2a-图2c所示。
3、线性范围的考察
精密称取巴巴酸对照品5.6mg,置10ml的容量瓶中,然后加5.6ml甲醇溶成1mg/ml的巴巴酸对照品溶液,精密吸取巴巴酸对照品溶液0.1ml、0.2ml、0.4ml、0.6ml、1ml分别置于10ml容量瓶中,加入甲醇溶液溶解稀释至刻度,配成10μg/ml、20μg/ml、40μg/ml、60μg/ml、100μg/ml,按上述色谱条件,进样测定,以巴巴酸对照品溶液的浓度(c,μg/ml)为横坐标,峰面积(a)为纵坐标进行线性回归,所测得的回归方程是y=34.509x-26.78(r=0.9993),结果在10μg/ml~100μg/ml之间与峰面积呈现较好的线性关系,如表1所示。色谱图如图4a-4f所示,图4a-4e分别表示浓度为10.0μg/ml、20μg/ml、40μg/ml、60μg/ml、100μg/ml的标准曲线实验图谱,图4f则为线性关系考察曲线示意图。
表1色谱条件下测定巴巴酸线性范围试验结果
4、精密度实验
精密吸取巴巴酸对照品溶液(40.0μg/ml)按上述色谱条件,进样测定,连续6次,按照测定的峰面积,计算出平均峰面积值为1321.0,rsd为0.3%,结果表明试验所用的色谱条件精密度良好,如表2所示。色谱图如图5a-图5f所示,图5a-图5f分别表示6次测定的实验图谱。
表2色谱条件下测定巴巴酸精密度试验结果
5、稳定性实验
精密吸取供试品溶液按上述色谱条件,分别于0h、2h、4h、6h、8h、10h注入高效液相色谱仪,测定不同时间段的峰面积,结果表明在10h内巴巴酸的平均峰面积为2279.6,rsd为0.2%(n=6),结果表明供试品溶液在10h之内稳定性良好,见表3。色谱图如图6a-6f所示。图6a-6f分别表示在0h、2h、4h、6h、8h、10h将供试品溶液注入高效液相色谱仪的实验图谱。
表3色谱条件下测定巴巴酸稳定性试验结果
6、重复性实验
取同一凝胶制剂6份,每份1g,精密称定,分别置于25ml的具塞锥形瓶中,精密加入20ml乙腈∶0.1%磷酸=80∶20(体积比)的溶液,称定重量,超声20min,放冷,再称定重量,用乙腈∶0.1%磷酸=80∶20(体积比)的溶液补足损失的重量,摇匀后,置50ml的离心管中,高速离心12min,取上清微孔滤膜滤过,按上述色谱条件,注入高效液相色谱仪中测定,以测得的峰面积计算含量,每克凝胶制剂含有巴巴酸1.29mg,rsd为2.90%(n=6),结果表明重现性好,结果如表4所示。色谱图如图7a-图7f所示,图7a-图7f分别表示6次实验的实验图谱。
表4色谱条件下测定巴巴酸重复性试验结果
7、加样回收率实验
称取已知含量的供试品(含量为1.29mg/g)6份,每份量为0.5g,精密称定,分别加入已知含量的巴巴酸对照品,置于25ml的具塞锥形瓶中,精密加入20ml的乙腈∶0.1%磷酸=80∶20(体积比)的溶液,称定重量,超声20min,放冷,称定重量,用乙腈∶0.1%磷酸=80∶20(体积比)的溶液补足重量,摇匀后,置50ml的离心管中,高速离心12min,取上清微孔滤膜滤过,按上述色谱条件,注入高效液相色谱仪中测定,计算回收率和rsd%值,平均回收率为99.36%,rsd为1.69%(n=6),结果如表5所示。色谱图如图8a-图8g所示,图8a所示为空白对照试验图谱,图8b-图8g分别表示试验1-6的实验图谱。
表5色谱条件下测定巴巴酸加样回收试验结果
8、样品的测定
精密称取10个不同批次的凝胶制剂各1g,分别置于25ml的具塞锥形瓶中,精密加入20ml的乙腈∶0.1%磷酸=80∶20(体积比)溶液,称定重量,超声20min,取出放冷,再称定重量,以乙腈∶0.1%磷酸=80∶20(体积比)的溶液补足重量,摇匀后,置50ml的离心管中,高速离心12min,取上清微孔滤膜滤过,即得供试品溶液。
分别取供试品溶液注入高效液相色谱仪测定,以峰面积分别计算10个样品巴巴酸含量,试验结果如表6所示。
表610批样品测定试验结果
对于本领域的技术人员来说,可以根据以上的技术方案和构思,作出各种相应的改变和变形,而所有的这些改变和变形都应该包括在本发明权利要求的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!