肽‑药物偶联物的制作方法
本发明涉及肿瘤特异性的肽-药物偶联物和包含所述偶联物的药物组合物。本发明还涉及此类偶联物和组合物作为抗肿瘤剂用于哺乳动物,特别是人的癌症治疗的用途。
背景和相关技术
与正常细胞相比,癌细胞常常过表达某些蛋白酶。这促进了对于靶向癌细胞的努力,通过将细胞毒性治疗剂与被肿瘤蛋白酶裂解的肽连接,以在癌细胞附近或内部释放细胞毒性药物同时不伤害或基本上不影响正常细胞。
美国专利号6,214,345公开了包括自切除接头并且在肿瘤的部位处选择性活化的肿瘤特异性肽-药物偶联物,其中所述药物可以是丝裂霉素或阿霉素,并且自切除接头是对-氨基苄醇。
表达天冬酰胺酶的细胞为肽-药物偶联物方法制造有吸引力的靶标,这是因为许多肿瘤过表达这些蛋白酶。一种这样的天冬酰胺酶,豆荚蛋白(legumain)在这方面吸引了特别的注意(Wu等人,Cancer R esearch 2006;66:970-980;Liu等人,Cancer Research 2003;63:2957–2964;Bajjuri等人,ChemMedChem.2011;6:.doi:10.1002/cmdc.201000478;http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3549592/pdf/nihm 54-59 s-340268.pdf.)。
丝裂霉素、阿霉素和喜树碱作为用于肽-药物偶联疗法的药物引已起了注意。美国专利号7,608,591;美国公开申请20110300147;美国公开申请20090175873;以及美国专利号8,314,060公开了用包含阿霉素的药物来靶向豆荚蛋白的肽-药物偶联物的实例。
上述参考文献中的每个的全文以引用方式并入本文。
这些先前的方法通常涉及药物部分的化学修饰,其可能不利地影响药物的功效。肽直接(通过C-末端羧基基团)与丝裂霉素的氮丙啶N原子偶联产生仲酰胺,其是通常不受蛋白酶攻击的官能团。此外,对于丝裂霉素的研究表明NH基团在氮丙啶环中对生物活性的作用。因此,存在对于将诸如丝裂霉素的抗肿瘤药物靶向表达豆荚蛋白和其他天冬酰胺酶的肿瘤细胞的改进偶联物的需要。
附图简述
图1是显示当暴露于人血浆时Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-丝裂霉素[本文的缀合物8]的衰变速率作为时间的函数的图。
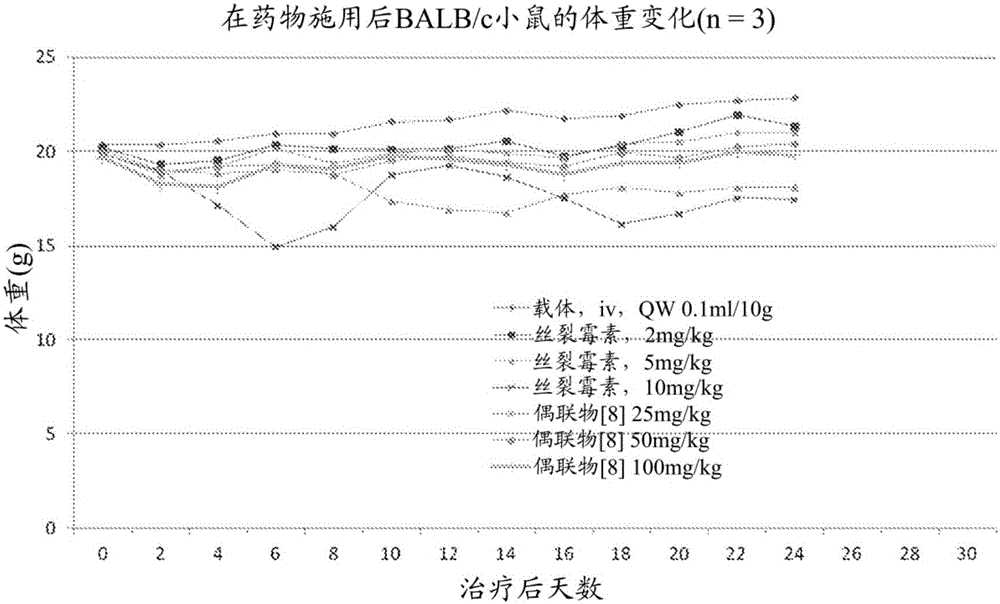
图2是显示在施用Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-丝裂霉素后Balb/c小鼠的体重作为时间的函数的图。
图3是显示在施用Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-丝裂霉素后Balb/c小鼠中的皮下CT-26同系结肠癌模型的肿瘤生长曲线的图。
技术实现要素:
本发明的某些实施方案是肽-药物偶联物。此类偶联物一般包括:1)可以被细胞蛋白酶裂解的肽部分,其结合2)自切除接头,特别是对-氨基苄基氨基甲酰基或对-氨基苄基碳酸酯部分,其接着结合3)细胞毒性药物部分。在这些实施方案中,接头部分附接到天冬酰胺残基,在所述位置发生蛋白水解裂解。
在特定实施方案中,肽-药物偶联物具有下式1的结构:
R-Y-Z-Asn-接头-D(式I)
其中
R包括选自由以下组成的组的取代基:
i)来源于C1至约C20直链、支链或脂环族羧酸的酰基、氨基甲酰基、磺酰基、磷酰基或烷基,其任选地被1至约5个羟基、胺基、羧基、磺酸基或磷酰基取代,
ii)具有1至约50个L-或D-氨基酸残基的肽,以及
iii)分子量为400至约40,000的聚乙二醇;
Y是选自由Ala、Thr、Ser、Leu、Arg、Pro、Val、Tyr、Phe组成的组的氨基酸残基;
Z是选自由Ala、Thr、Asn和Pro组成的组的氨基酸残基;
Asn是天冬酰胺残基;
接头是对-氨基苄基氨基甲酰基部分或对-氨基苄基碳酸酯部分;
D是结合到接头部分的抗肿瘤药物部分,其中所述药物选自由以下组成的组:丝裂霉素、阿霉素、氨基蝶呤、放线菌素、博来霉素、9-氨基-喜树碱、N8-乙酰基亚精胺、1-(2-氯乙基)-1,2-二甲磺酰肼、他利霉素、阿糖胞苷、依托泊苷、喜树碱、紫杉醇、拉霉素、鬼臼毒素、蛇形菌素(anguidine)、长春新碱、长春碱、吗啉-阿霉素、正-(5,5-二乙酰氧基-戊基)阿霉素及其衍生物。
在式I的特定实施方案中:
R包括选自由以下组成的组的取代基:
i)来源于C1至约C20直链、支链或脂环族羧酸的酰基,其任选地被1至约5个羟基、胺基、羧基、磺酸基或磷酰基取代,
ii)具有1至约50个L-氨基酸残基的肽,以及
iii)分子量为400至约40,000的聚乙二醇;
Y是选自由Ala、Thr、Ser、Leu、Arg、Pro、Val、Tyr、Phe组成的组的氨基酸残基;
Z是选自由Ala、Thr、Asn和Pro组成的组的氨基酸残基;
Asn是天冬酰胺残基;
接头是对-氨基苄基氨基甲酰基部分;
D是抗肿瘤药物部分,其中所述药物选自由丝裂霉素和阿霉素组成的组。
在式I的其他特定实施方案中:
R包括选自由以下组成的组的取代基:
i)来源于C1至约C20直链、支链或脂环族羧酸的酰基、氨基甲酰基、磺酰基、磷酰基或烷基,其任选地被1至约5个羟基、胺基、羧基、磺酸基或磷酰基取代,
ii)具有1至约50个L-或D-氨基酸残基的肽,以及
iii)分子量为400至约40,000的聚乙二醇;
Y是选自由Ala、Thr、Ser、Leu、Arg、Pro、Val、Tyr、Phe组成的组的氨基酸残基;
Z是选自由Ala、Thr、Asn和Pro组成的组的氨基酸残基;
Asn是天冬酰胺残基;
接头是对-氨基苄基碳酸酯部分;并且
D是结合到接头部分的抗肿瘤药物部分,并且其中所述药物是喜树碱。
在仍然进一步的实施方案中,本发明包括式II所示的式的二聚肽-药物偶联物:
D’-接头-Asn-Z-Y-R’-Y-Z-Asn-接头-D(式II)
其中D和D’是彼此相同、不同的细胞毒性药物部分,并且独立地选自由以下组成的组:丝裂霉素、阿霉素、氨基蝶呤、放线菌素、博来霉素、9-氨基-喜树碱、N8-乙酰基亚精胺、1-(2-氯乙基)-1,2-二甲磺酰肼、他利霉素、阿糖胞苷、依托泊苷、喜树碱、紫杉醇、拉霉素、鬼臼毒素、蛇形菌素、长春新碱、长春碱、吗啉-阿霉素、正-(5,5-二乙酰氧基-戊基)阿霉素及其衍生物;
接头是对-氨基苄基氨基甲酸酯部分或对-氨基苄基碳酸酯部分,其中所述接头可以是相同或不同的;
R’包括选自由以下组成的组的取代基:
i)选自由任选地被1至约5个羟基、胺基、羧基、磺酸基或磷酰基取代的来源于C1至约C20直链、支链或脂环族羧酸的酰基、氨基甲酰基、磺酰基、磷酰基或烷基组成的组的双官能团,
ii)具有1至约50个L-或D-氨基酸残基的肽,以及
iii)分子量为400至约40,000的聚乙二醇;并且
Y是选自由Ala、Thr、Ser、Leu、Arg、Pro、Val、Tyr、Phe组成的组的氨基酸残基;
Asn是天冬酰胺残基;并且
Z是选自由Ala、Thr、Asn和Pro组成的组的氨基酸残基。
如将理解的,式II的偶联物基本上是式I的偶联物的二聚体形式,其可以使每个偶联物的两个细胞毒性药物分子靶向靶细胞或组织。
在进一步的实施方案中,本发明包括一种药物组合物,其在至少一种药学上可接受的载体中包含至少一种式I或式II的肽-药物偶联物。
在仍然进一步的实施方案中,本发明包括一种治疗哺乳动物(其可以是人)的癌症的方法,其包括施用抗肿瘤有效量的本发明偶联物或药物组合物。
发明详述
如此处所用的“丝裂霉素”是指从头状链霉菌(Streptomyces caespitosus)或淡紫灰链霉菌(Streptomyces lavendulae)分离的含氮丙啶的药物家族的成员,并且特别包括丝裂霉素C和丝裂霉素A。
如本文所用的“阿霉素”是指来源于链霉菌细菌波赛链霉菌(Streptomyces peucetius var.caesius)的蒽环类家族的成员,并且包括阿霉素、柔红霉素、表柔比星和伊达比星。
如此处所用的“喜树碱”是指从喜树(Camptotheca acuminata)及其化学衍生物分离的生物碱家族的成员,并且包括喜树碱、伊立替康、拓扑替康和卢比替康。
本发明提供肿瘤特异性的肽-药物偶联物,其包括肽部分、自切除接头和细胞毒性药物部分,所述自切除接头为对氨基苄基-氨基甲酰基或对-氨基苄基碳酸酯(取决于包含在自切除接头所附接的药物中的官能团的类型)。在偶联物基本上无活性和无毒性的意义上,所述偶联物用作前药。肽部分可以被蛋白酶在体内选择性地裂解以释放自切除接头/细胞毒性药物部分。在这种酶裂解后,自切除接头自发水解以产生为其活性形式,但是更加直接针对靶向环境(诸如人类患者的肿瘤部位)的游离药物。以这种方式,细胞毒性药物靶向需要治疗的特定部位,而减少除靶标部位以外的部位处的细胞和组织损伤。
细胞毒性药物部分具有化学反应性官能团,药物主链通过所述化学反应性官能团共价键合到自切除接头。将细胞毒性药物连接到自切除接头的官能团使得在自切除接头水解时,细胞毒性药物以细胞毒性活化形式被释放。这种官能团可包括,例如伯胺、仲胺或羟基。细胞毒性药物包括丝裂霉素、阿霉素、氨基蝶呤、放线菌素、博来霉素、9-氨基-喜树碱、N8-乙酰基亚精胺、1-(2-氯乙基)-1,2-二甲磺酰肼、他利霉素、阿糖胞苷、依托泊苷、喜树碱、紫杉醇、拉霉素、鬼臼毒素、蛇形菌素、长春新碱、长春碱、吗啉-阿霉素、正-(5,5-二乙酰氧基-戊基)阿霉素及其衍生物。优选的实施方案是基于丝裂霉素、阿霉素和/或喜树碱的。
在药物(D)是丝裂霉素的具体实施方案中,肽-药物偶联物具有如式III所示的结构(其中R如上所定义,并且Ala是丙氨酸残基):
式III
在药物(D)是阿霉素的具体实施方案中,肽-药物偶联物具有如式IV所示的结构(其中R如上所定义,并且Ala是丙氨酸残基):
式IV
在药物(D)是喜树碱的具体实施方案中,肽-药物偶联物具有如式V所示的结构(其中R如上所定义,并且Ala是丙氨酸残基):
式V
在药物(D)是丝裂霉素且药物(D’)是阿霉素的二聚偶联物的具体实施方案中,肽-药物偶联物具有如式VI所示的结构(其中R'如上所定义,并且Ala是丙氨酸残基):
式VI
在药物(D)是丝裂霉素且药物(D')是喜树碱的二聚偶联物的具体实施方案中,肽-药物偶联物具有如式VII所示的结构(其中R'如上定义,并且Ala是丙氨酸残基):
式VII
本发明的优选偶联物包括:
Ala-Ala-Asn-PABC-丝裂霉素:
Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-丝裂霉素:
Nα-乙酰胺-Ala-Ala-Asn-PABC-丝裂霉素;
Nα-丁酰胺-Ala-Ala-Asn-PABC-丝裂霉素;
Nα-己酰胺-Ala-Ala-Asn-PABC-丝裂霉素;
Nα-[-(2-酰胺-2-氧代乙氧基)乙酸]-Ala-Ala-Asn-PABC-丝裂霉素;以及
Nα-[-((2-酰胺-2-氧代乙氧基)(甲基)氨基)乙酸]-Ala-Ala-Asn-PABC-丝裂霉素;Ala-Ala-Asn-PABC-阿霉素;
Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-阿霉素;
Nα-乙酰胺-Ala-Ala-Asn-PABC-阿霉素;
Nα-丁酰胺-Ala-Ala-Asn-PABC-阿霉素;以及
Nα-己酰胺-Ala-Ala-Asn-PABC-阿霉素;
Nα-[-(2-酰胺-2-氧代乙氧基)乙酸]-Ala-Ala-Asn-PABC-阿霉素;以及
Nα-[-((2-酰胺-2-氧代乙氧基)(甲基)氨基)乙酸]-Ala-Ala-Asn-PABC-阿霉素;其中PABC是对-氨基苄基氨基甲酰基接头部分。
本发明的其他优选偶联物包括:
Ala-Ala-Asn-PABC-喜树碱;
Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-喜树碱;
Nα-[-(2-酰胺-2-氧代乙氧基)乙酸]-Ala-Ala-Asn-PABC-喜树碱;以及
Nα-[-((2-酰胺-2-氧代乙氧基)(甲基)氨基)乙酸]-Ala-Ala-Asn-PABC-喜树碱
其中PABC是对-氨基苄基碳酸酯部分。
优选的二聚偶联物包括:
N1-Ala-Ala-Asn-PABC-丝裂霉素、N4-Ala-Ala-Asn-PABC-阿霉素–琥琥珀酰胺:
N1-Ala-Ala-Asn-PABC-丝裂霉素、N5-Ala-Ala-Asn-PABC-阿霉素-双(Oα)-乙酰胺:
N1-Ala-Ala-Asn-PABC-丝裂霉素、N5-Ala-Ala-Asn-PABC-阿霉素-双(Nα-甲基)-乙酰胺;
N1-Ala-Ala-Asn-PABC-丝裂霉素、N4-Ala-Ala-Asn-PABC-喜树碱–琥珀酰胺;
N1-Ala-Ala-Asn-PABC-丝裂霉素、N5-Ala-Ala-Asn-PABC-喜树碱-双(Oα)-乙酰胺;以及
N1-Ala-Ala-Asn-PABC-丝裂霉素、N5-Ala-Ala-Asn-PABC-喜树碱-双(Nα-甲基)-乙酰胺。
其中PABC是对-氨基苄基氨基甲酰基或对-氨基苄基碳酸酯接头部分。
肽-药物偶联物的制备
通常,式I和式II的肽-药物偶联物可使用可用材料和常规有机合成技术来制备。例如,所述类型的细胞毒性药物是可商购获得的,并且它们的合成描述于科学文献中。
通常,本发明的肽-药物偶联物可通过将药物部分通过自切除接头共价附接到肽序列来构建。下面所示的制备的具体合成路线是可使用的那些的示例。
合成方案1示出用于生产肽-丝裂霉素偶联物的合成路线:
合成方案1
合成方案2示出用于生产肽-阿霉素偶联物的合成路线:
合成方案2
合成方案3示出用于生产肽-喜树碱偶联物的合成路线:
合成方案3
合成方案4示出用于生产阿霉素-肽-丝裂霉素偶联物(二聚偶联物)的合成路线:
合成方案4
合成方案5示出用于生产喜树碱-肽-丝裂霉素偶联物(二聚偶联物)的合成路线:
合成方案5
通常对于所述的合成反应:
a)采用标准肽合成方法用于Fmoc和三苯甲基迁放和肽偶联;
b)通过在有机/水性溶剂混合物中使用EEDQ作为偶联试剂,通过使Nα-Fmoc-Asn或三苯甲基保护的Nα-Fmoc-Asn与对-氨基苄醇反应来使自切除接头附接到氨基酸;
c)可以通过使对-硝基苯氯甲酸酯与Nα-Fmoc-Asn-PAB-OH或Nα-Fmoc-Ala-Ala–N-三苯甲基-Asn-PAB-OH反应来获得对-氨基苄醇的活化碳酸酯;
d)在HOBT存在下在DMF中,通过与Nα-Fmoc-Asn-PABC-PNP或Nα-Fmoc-Ala-Ala-Asn-PABC-PNP的相应活化碳酸酯反应来获得丝裂霉素和阿霉素的肽药物偶联物;
e)对于喜树碱偶联物的合成,使喜树碱与三光气反应以原位提供喜树碱氯甲酸酯,然后与Nα-Fmoc-Asn-PAB-OH偶联以获得相应的Nα-Fmoc-Asn-PABC-喜树碱;
f)通过肽偶联方法与各种酸酐、酰氯或羧酸酰化来获得最终的肽-药物偶联物。
合成的最终偶联物可以通过各种方法来纯化,包括二氧化硅柱色谱、HPLC、离子交换色谱、酸/碱沉淀和结晶。
最终的偶联物可通过1H-NMR、13C-NMR、MS、LC/MS、UV/VIS和/或IR来表征。
许多所公开的偶联物可以作为盐酸盐或其他盐存在。药物化学领域的技术人员将理解,盐的选择不是关键的,并且其他药学上可接受的盐可以通过熟知的方法制备且可以用于制备药物组合物。参见例如,Handbook of Pharmaceutical Salts:Properties,Selection and Use.(P.Heinrich Stahl和Camille G.Wermuth编辑)International Union of Pure and Applied Chemistry,Wiley-VCH 2002 and L.D.Bighley,S.M.Berge,D.C.Monkhouse,in"Encyclopedia of Pharmaceutical Technology'.编辑J.Swarbrick和J.C.Boylan,第13卷,Marcel Dekker公司,New York,Basel,Hong Kong 1995,第453-499页。
此外,本领域的技术人员将理解,不仅可以产生和使用多种盐,而且可以从本文公开的偶联物产生水合物、溶剂合物和多晶型物。此外,也可以容易地产生各种同位素取代的变体(通过例如,氘代替氢、13C代替碳、15N代替氮)。此类衍生物被考虑在本公开的范围内。
为了制备本发明的药物组合物,将一种或多种偶联物与至少一种药学上可接受的载体相结合。“药学上可接受的载体”是指适用于药理学有效物质的特定施用途径的生物相容性化合物。它们包括稳定剂、润湿剂和乳化剂、用于改变渗透压的盐、包封剂、缓冲剂以及皮肤渗透促进剂。药学上可接受的载体的实例描述于Remington的药物科学(Alfonso R.Gennaro编辑,第18版,1990)中。一种载体或多种载体的特定选择取决于预期的具体治疗。用于药物组合物及其组分的各种制剂描述于美国公开专利申请2014/0057844中,其公开内容通过引用并入本文。
偶联物中细胞毒性药物部分的选择由待治疗的癌症的类型来指导。对于特定类型的癌症或肿瘤的治疗,细胞毒性药物部分应基于有效治疗此类型癌症的细胞毒性药物。例如,基于丝裂霉素的药物偶联物可用于根据本领域目前已知和推荐的丝裂霉素施用方案或由其指导来治疗癌症。
本发明的偶联物、组合物和方法可用于治疗不同类型的癌症,包括但不限于膀胱癌、乳腺癌、子宫颈癌、卵巢癌、胃癌、胰腺癌、肺癌、肝癌、食道癌、肠癌、皮肤癌以及前列腺癌。
施用途径包括注射、口服、经颊施用、肠胃外施用、吸入以及直肠施用。
待施用的偶联物的剂量以及施用的具体途径和方案取决于待治疗的癌症类型和特定癌症病状的情况,但可以由本领域技术人员确定。
本发明的偶联物可彼此组合使用并且与其他化疗剂或治疗组合使用。例如,使用本发明偶联物的治疗可与放射治疗组合使用。
下面的实施例说明了本发明的某些实施方案,并且应被认为是说明性的而不是限制本发明的范围。
实施例
生物活性
在体外和体内两种系统中测试本发明的代表性肽-药物偶联物以确定其生物活性。在这些测试中,通过测量偶联物针对人癌症来源的细胞的细胞毒性来确定细胞毒性药物的偶联物的效力。本领域技术人员将认识到,可以使用表达期望的肿瘤相关蛋白酶(裂解本发明的偶联物以释放为活性形式的药物的蛋白酶)的任何肿瘤细胞系代替在下面分析中使用的特定肿瘤细胞系。下面描述使用的代表性测试和获得的结果。
测试I
人血浆稳定性
用人血浆将20μL在DMSO储备溶液中的500μM肽-药物偶联物Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-丝裂霉素[本文的化合物8]稀释至1mL(最终浓度:10μM,2%DMSO),并且将混合物在37℃下孵育。在0、0.25、0.5、1、2、4、6小时的时间点移除100μL等分试样,并且用含有甲苯磺丁脲(200ng/mL)作为内标的400μL冷乙腈稀释。将样品在14,000rpm下离心4分钟。用300μL 0.1%甲酸HPLC水将100μL上述上清液稀释,并且取10μL用于LC/MS/MS分析(柱:C-18;流动相:0.1%甲酸的水溶液/0.1%甲酸的乙腈溶液;离子跃迁:Q1离子(m/z)=840.5,Q2离子(m/z)=462.2)。如图1所示,肽-药物偶联物在人血浆中相对稳定,其中半衰期大于15小时(T1/2>15h)。检测到少于2%的游离药物丝裂霉素。
测试II
体外细胞毒性测定
使用胰蛋白酶-EDTA收获人癌细胞的单层培养物,并且将细胞计数并且在含有10%FBS的RPMI-1640或DMEM中重悬浮至1x105/mL。将细胞(0.1mL/孔)添加到96孔微量滴定板的每个孔中,并且在37℃下,在5%CO2的潮湿气氛中孵育过夜。将培养基从板上移除,并且将丝裂霉素或偶联物在培养基中的系列稀释液(最终DMSO浓度<0.1%)添加到孔中。一式三份进行所有稀释。将药物处理的细胞在37℃下,在5%CO2的潮湿气氛中再培养72小时。向每个孔中添加50μL冷TCA(50%,重量/体积),并且将板在4℃下孵育1小时。用缓慢流动的自来水将板洗涤三次,并且在室温下干燥。向每个孔中添加50μL磺酰罗丹明B溶液(0.4%,重量/体积),并且将板在室温下放置1小时。用乙酸溶液(1%,体积/体积)冲洗板以去除未结合的染料并且在室温下干燥。向每个孔中添加200μL的10mM Tris碱溶液,并且将板在回转振荡器上摇动15分钟以溶解蛋白结合染料。在微板读取器中测量510nm处的孔光密度,并且通过GraphPad从每个实验中三次重复的三个分离实验来计算IC50,并且表示为平均值(表1)。
表1 IC50(μM,n=3)
HCT116和HepG2人癌细胞系测定显示,与母体药物丝裂霉素相比,肽-药物偶联物的细胞毒性降低了76至17倍,这取决于不同的肿瘤细胞系。在缺氧和酸性条件下,肿瘤相关蛋白质豆荚蛋白在实体瘤的肿瘤微环境中过表达。然而,先前报道了细胞培养物中HCT116和HepG2两种细胞系的豆荚蛋白表达的一定水平,其可导致如在此测定中观察到的肽-药物偶联物的残留活性。
测试III
Balb/c小鼠中的体内最大耐受剂量(MTD)
在Balb/c小鼠中分别评估丝裂霉素及其肽-药物偶联物Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-丝裂霉素[化合物8]作为单一药剂的耐受性。在治疗后不同时间点处的不同组的体重结果如图2所示。
在10mg/kg,每周一次丝裂霉素的剂量组中,观察到动物死亡。在5mg/kg组中,小鼠显示立毛和迟钝的行为。在5mg/kg丝裂霉素组中观察到体重减轻(>15%)。同时,在25、50和100mg/kg,每周一次,共三次注射持续24天的剂量组中,对于肽-药物偶联物Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-丝裂霉素[8],没有观察到异常的外观和体重(<10%)。鼠毒性研究显示,肽-药物偶联物Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-丝裂霉素[8]在体内比母体药物丝裂霉素毒性小得多。与母体药物丝裂霉素相比,肽-药物偶联物的小鼠最大耐受剂量(MTD)增加至少20倍。
测试IV
体内抗肿瘤活性
在Balb/c小鼠的皮下CT-26同系结肠癌模型上评估肽-药物偶联物Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-丝裂霉素[8]的杀肿瘤效果。每只小鼠在右侧腹部皮下接种在0.1mL PBS中的CT-26肿瘤细胞(5x105)。当平均肿瘤尺寸达到大约180mm3(约10天后)时,通过静脉内施用:载体(2%DMA和98%的40%2HP-β-CD)、丝裂霉素(2mg/kg;5mg/kg,QW)和偶联物Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-丝裂霉素[8](25mg/kg;50mg/kg,QW)持续三周,每只小鼠共注射三次来治疗CT-26肿瘤小鼠。CT-26模型的肿瘤生长曲线如图3所示。
如先前报道的,即使CT-26细胞在体外具有肿瘤相关蛋白酶豆荚蛋白的弱表达(week expression),其也在体内在CT-26实体瘤微环境中的活的内皮细胞和肿瘤相关巨噬细胞表面上的TME中大量表达,这是因为豆荚蛋白表达是在缺氧和应激条件下被诱导的。豆荚蛋白特异性活化偶联物Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-丝裂霉素[8]在Balb/c小鼠的皮下CT-26合成结肠癌模型上表现出强的抗肿瘤功效,如图3所示。在比其MTD低得多的50mg/kg剂量下,与未治疗的对照相比,偶联物显著抑制肿瘤生长(T/C=36.4%,p<0.01)。虽然在其MTD剂量(5mg/kg)处,母体药物丝裂霉素表现出更弱的肿瘤生长抑制效果(T/C=50.4%)。因此,体内实验表明,本发明的肽-丝裂霉素偶联物对宿主产生具有比母体药物丝裂霉素更大效力和更低毒性的抗肿瘤活性。
合成实施例
实施例1
制备Nα-Fmoc-Ala-Ala[1]
将在CH2Cl2(50mL)中的Nα-Fmoc-Ala(1.58g,5.0毫摩尔)、N-羟基琥珀酰亚胺(0.63g,5.5毫摩尔)和DCC(1.03g,5.0毫摩尔)的溶液在5℃下搅拌6小时。将DCU滤出,并且将滤液浓缩。将残余物再溶解于THF(50mL)中并且在冰箱(4℃)中保持过夜。滤出更多的DCU,并且将THF溶液添加到在25%THF/H2O(80mL)中的丙氨酸(0.99g,7.5毫摩尔)和NaHCO3(1.68g,20毫摩尔)的溶液中。将反应混合物在25℃下剧烈搅拌5小时。通过浓缩去除THF,并且用浓HCl将水性悬浮液调节至pH4。将水性悬浮液在25℃下再搅拌3小时,并且通过过滤收集沉淀物,用去盐水充分冲洗,并且在真空中用KOH干燥,得到白色固体产物(1.77g,83.7%产率)。
LC/MS:(MH)+=383
实施例2
制备Nα-Fmoc-Asn-PAB-OH[2]
将在THF/H2O(100/20mL)中的Nα-Fmoc-Asn(1.77g,5.0毫摩尔)、对-氨基苄醇(0.86g,7.0毫摩尔)和EEDQ(1.48g,6毫摩尔)的溶液在室温下搅拌过夜。添加另外量的EEDQ(0.61g,2.5毫摩尔),并且再搅拌24小时。通过浓缩去除THF,并且用NaOH/NaHCO3水溶液(2/8g,200mL)稀释残余物悬浮液,并且搅拌3小时。通过过滤收集沉淀物,用水冲洗并且再悬浮于10%柠檬酸(150mL)中。通过过滤收集沉淀物,用10%柠檬酸随后用去盐水冲洗,并且在真空中干燥。将上面获得的固体在乙酸乙酯(100mL)中研磨。通过过滤收集固体并且在真空中干燥,得到灰白色产物(0.95g,40%产率)。
LC/MS:(MH)+=460
实施例3
制备Nα-Fmoc-Asn-PABC-PNP[3]
在室温下,用对-硝基苯氯甲酸酯(0.4g,2.0毫摩尔)和吡啶(0.15g,2.0毫摩尔)处理在干燥THF/DMF(50/5mL)中的Nα-Fmoc-Asn-PAB-OH[2](0.75g,1.6毫摩尔)。16小时后,添加另外量的对-硝基苯氯甲酸酯(0.2g,1.0毫摩尔),并且将反应溶液再搅拌6小时。用乙酸乙酯(250mL)稀释上述溶液,并且用5%柠檬酸(2x100mL)随后用盐水洗涤,用Na2SO4干燥,并且蒸发至干燥。将残余物在50%EA/己烷中研磨,并且通过过滤收集固体,得到灰黄色(off-yellow)产物(0.8g,81%产率)。
LC/MS:(MH)+=625
实施例4
制备Nα-Fmoc-Asn-PABC-丝裂霉素[4]
在室温下,用HOBT(675mg,5.0毫摩尔)和DIEA(650mg,5.0毫摩尔)将在干燥DMF(15mL)中的Nα-Fmoc-Asn-PABC-PNP[3](624mg,1.0毫摩尔)和丝裂霉素(400mg,1.2毫摩尔)处理4小时。用乙酸乙酯(150mL)稀释反应混合物,并且用NaOH/NaHCO3(1/4g,200mL)随后用盐水洗涤三次。用Na2SO4干燥有机层并且在减压下浓缩至干燥。将残余物在50%乙酸乙酯/己烷(50mL)中研磨,并且通过过滤收集固体,用乙酸乙酯/己烷冲洗并且在真空中干燥,得到紫色产物(635mg,76.3%产率)。
LC/MS:(MH)+=820
实施例5
制备Asn-PABC-丝裂霉素[5]
在室温下用20%吗啉/NMP(15mL)处理Nα-Fmoc-Asn-PABC-丝裂霉素[4](624mg,0.76毫摩尔)。30分钟后,添加50%乙酸乙酯/己烷(100mL),并且去除上清液。将上述过程重复两次。将残余物再溶解于甲醇(25mL)中,并且将溶剂在减压下蒸发至干燥。将残余物在50%乙酸乙酯/己烷(100mL)中研磨并且在室温下搅拌过夜。通过过滤收集固体,用乙酸乙酯/己烷冲洗并且在真空中干燥,得到紫色产物(440mg,95.6%产率)。
LC/MS:(MH)+=598
实施例6
制备Nα-Fmoc-Ala-Ala-Asn-PABC-丝裂霉素[6]
在室温下用PyBop(390mg,0.75毫摩尔)和DIEA(585mg,4.5毫摩尔)处理在NMP(15mL)中的Asn-PABC-丝裂霉素[5](440mg,0.73毫摩尔)和Nα-Fmoc-Ala-Ala[1](286mg,0.75毫摩尔)。1小时后,用乙酸乙酯(200mL)稀释反应混合物。用5%柠檬酸(3x100mL)、盐水洗涤有机溶液,并且用Na2SO4干燥。在减压下蒸发溶剂,并且将残余物在50%乙酸乙酯/己烷(100mL)中研磨,并且在室温下搅拌过夜。通过过滤收集固体,并且再悬浮于乙酸乙酯(50mL)中并超声处理。通过过滤收集固体,用乙酸乙酯充分冲洗并且在真空中干燥,得到紫色产物(530mg,75.4%产率)。
LC/MS:(MH)+=962
实施例7
制备Ala-Ala-Asn-PABC-丝裂霉素[7]
在室温下在20%吗啉/NMP(10mL)中处理Nα-Fmoc-Ala-Ala-Asn-PABC-丝裂霉素[6](481mg,0.5毫摩尔)。30分钟后,添加50%乙酸乙酯/己烷(150mL),并且搅拌1小时。去除上清液,并且将残余物在CH2Cl2(50mL)中研磨。通过过滤收集固体,并且再溶解于甲醇(20mL)中。在减压下蒸发溶剂,并且将残余物在乙酸乙酯中研磨。通过过滤收集固体,用乙酸乙酯充分冲洗并且在真空中干燥,得到紫色产物(277mg,75%产率)。
LC/MS:(MH)+=740
实施例8
制备Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-丝裂霉素[8]
在室温下,用琥珀酸酐(50mg,0.5毫摩尔)和DIEA(130mg,1.0毫摩尔)处理在DMA/CH2Cl2(2/6mL)中的Ala-Ala-Asn-PABC-丝裂霉素[7](260mg,0.35毫摩尔)。将反应混合物搅拌过夜。向上述反应混合物中添加乙醚(100mL)并且搅拌30分钟。去除上清液,并且将所述过程重复两次。将残余物在2%HOAc/乙酸乙酯(50mL)中研磨,并且通过过滤收集固体,用乙酸乙酯充分冲洗并且在真空中干燥,得到紫色产物(265mg,90%产率)。
LC/MC:(M-H)–=838
实施例9
制备Nα-乙酰胺-Ala-Ala-Asn-PABC-丝裂霉素[9]
在室温下,用乙酸酐(10mg,0.1毫摩尔)和DIEA(13mg,0.1毫摩尔)处理在DMA/CH2Cl2(1/3mL)中的Ala-Ala-Asn-PABC-丝裂霉素[7](37mg,0.05毫摩尔)。30分钟后,添加乙醚(50mL),并且搅拌60分钟。通过过滤收集软固体,用乙醚冲洗并且再溶解于甲醇(5mL)中。在减压下去除溶剂,并且将残余物在50%乙酸乙酯/己烷(10mL)中研磨,并通过过滤收集固体,用乙酸乙酯/己烷充分冲洗,在真空中干燥,得到紫色产物(35mg,90%产率)。
LC/MC:(MH)+=780;(M+Na)+=805
实施例10
制备Nα-丁酰胺-Ala-Ala-Asn-PABC-丝裂霉素[10]
在室温下,用丁酰氯(6.3mg,0.06毫摩尔)和DIEA(13mg,0.1毫摩尔)处理在DMA/CH2Cl2(1/3mL)中的Ala-Ala-Asn-PABC-丝裂霉素[7](37mg,0.05毫摩尔)。30分钟后,添加乙醚(50mL),并且搅拌60分钟。通过过滤收集固体,用乙醚冲洗并且再溶解于甲醇(5mL)中。在减压下去除溶剂,并且将残余物在乙酸乙酯(10mL)中研磨。通过过滤收集固体,用乙酸乙酯充分冲洗并且在真空中干燥,得到紫色产物(27mg,67%产率)。
LC/MC:(MH)+=808;(M+Na)+=833。
实施例11
制备Nα-己酰胺-Ala-Ala-Asn-PABC丝裂霉素[11]
在室温下,用己酰氯(9.0mg,0.06毫摩尔)和DIEA(13mg,0.1毫摩尔)处理在DMA/CH2Cl2(1/3mL)中的Ala-Ala-Asn-PABC-丝裂霉素[7](37mg,0.05毫摩尔)。30分钟后,添加乙醚(50mL),并且搅拌60分钟。通过过滤收集固体,用乙醚冲洗并且再溶解于甲醇(5mL)中。在减压下去除溶剂,并且将残余物在50%乙酸乙酯/己烷(10mL)中研磨。通过过滤收集固体,用乙酸乙酯充分冲洗并且在真空中干燥,得到紫色产物(27mg,67%产率)。
LC/MC:(MH)+=836;(M+Na)+=860
实施例12
制备Nα-[-(2-酰胺-2-氧代乙氧基)乙酸]-Ala-Ala-Asn-PABC-丝裂霉素[12]
在室温下用1,4-二噁烷-2,6-二酮(70mg,0.6毫摩尔)和DIEA(60mg,0.5毫摩尔)将在THF/DMF(4/0.5mL)中的Ala-Ala-Asn-PANC-丝裂霉素[7](295mg,0.4毫摩尔)处理2小时。缓慢添加乙醚(10mL),并且将混合物搅拌30分钟。去除溶剂,并且将残余物研磨并在乙酸乙酯中的1%乙酸中超声处理。通过过滤收集固体,用乙酸乙酯充分冲洗并且在真空中干燥,得到暗色产物(276mg,80%产率)。
LC/MC:(M-H)-=854
实施例13
制备Nα-[-((2-酰胺-2-氧代乙氧基)(甲基)氨基)乙酸]-Ala-Ala-Asn-PABC-丝裂霉素[13]
在室温下用4-甲基吗啉-2,6-二酮(77mg,0.6毫摩尔)和DIEA(130mg,1.0毫摩尔)将在THF/DMF(4/0.5mL)中的Ala-Ala-Asn-PANC-丝裂霉素[7](295mg,0.4毫摩尔)处理2小时。缓慢添加乙醚(10mL),并且将混合物搅拌30分钟。去除溶剂,并且将残余物研磨并在乙酸乙酯中的1%乙酸中超声处理。通过过滤收集固体,用乙酸乙酯充分冲洗并且在真空中干燥,得到暗色产物(339mg,97%产率)。
LC/MS:(M-H)-=867
实施例14
制备Nα-Fmoc-Asn(N-三苯甲基)-PAB-OH[14]
将在THF(50mL)中的Nα-Fmoc-Asn(N-三苯甲基)((1.49g,2.5毫摩尔)、对-氨基苄醇(0.37g,3.0毫摩尔)和EEDQ(0.74g,3毫摩尔)的溶液在室温下搅拌过夜。添加另外量的EEDQ(0.25g,1.0毫摩尔),并且再搅拌6小时。用乙酸乙酯(300mL)稀释反应溶液,用0.1N HCl洗涤三次,并且用Na2SO4干燥。通过短硅胶塞过滤乙酸盐溶液,用乙酸乙酯冲洗并且浓缩至干燥。将残余物在乙醚中研磨,过滤,用乙醚冲洗并且在真空中干燥,得到白色固体(1.65g,93%产率)。
LC/MS:(MH)+=702
实施例15
制备Asn(N-三苯甲基)-PAB-OH[15]
向在CH2Cl2(50mL)中的Nα-Fmoc-Asn(N-三苯甲基)-PAB-OH[14](1.6g,2.28毫摩尔)的溶液中添加DBU(1mL)。将反应溶液搅拌15分钟。用CH2Cl2(200mL)稀释反应溶液,用盐水洗涤两次,并且用Na2SO4干燥,过滤并且浓缩至干燥。将残余物在己烷/乙醚(1/1)中研磨。通过过滤收集固体,用乙醚冲洗并且在真空中干燥,得到白色产物(1.1g,100%产率)。
LC/MS:(MH)+=480
实施例16
制备Nα-Fmoc-Ala-Ala-Asn(N-三苯甲基)-PAB-OH[16]
将在NMP(20mL)中的Asn(N-三苯甲基)-PAB-OH[15](0.72g,1.5毫摩尔)、Nα-Fmoc-Ala-Ala[1](0.57g,1.5毫摩尔)、PyBop(0.94g,1.8毫摩尔)和DIEA(1.17g,9.0毫摩尔)的溶液在室温下搅拌1小时。在冰-水温度下向上述反应中缓慢添加5%柠檬酸水溶液(150mL),并且搅拌2小时。通过过滤收集沉淀物,并且用5%柠檬酸溶液随后用去盐水冲洗。将固体再悬浮于NaHCO3水溶液中,研磨,过滤,用去盐水冲洗并且在真空中用KOH干燥,得到白色产物(1.1g,87%产率)。
LC/MS:(MH)+=845
实施例17
制备Nα-Fmoc-Ala-Ala-Asn(N-三苯甲基)-PABC-PNP[17]
将在干燥THF中的Nα-Fmoc-Ala-Ala-Asn(N-三苯甲基)-PAB-OH[16](1.1g,1.3毫摩尔)、对-硝基苯氯甲酸酯(0.31g,1.6毫摩尔)和吡啶(0.13g,1.6毫摩尔)的溶液在室温下搅拌过夜。添加另外量的对-硝基苯氯甲酸酯(0.2g,1.0毫摩尔)和吡啶(0.08g,1.0毫摩尔),并且再搅拌4小时。用乙酸乙酯(300mL)稀释上述溶液,并且用5%柠檬酸溶液(3x100mL)随后用盐水洗涤,用Na2SO4干燥,过滤并且浓缩至干燥。将残余物在乙醚中研磨,过滤,用乙醚冲洗并且在真空中干燥,得到灰白色产物(1.1g,83%产率)。
LC/MS:(MH)+=1010
实施例18
制备Nα-Fmoc-Ala-Ala-Asn-PABC-PNP[18]
将在TFA/CH2Cl2(6/24mL)中的Nα-Fmoc-Ala-Ala-Asn(N-三苯甲基)-PABC-PNP[17](1.0g,1毫摩尔)和TIPS(2.5mL)的溶液在室温下搅拌2小时。向上述反应溶液中缓慢添加乙醚(150mL)并且将悬浮液搅拌1小时。通过过滤收集沉淀物并且用乙醚冲洗。将固体在乙酸乙酯中研磨,过滤,用乙酸乙酯冲洗并且在真空中干燥,得到灰白色产物(0.75g,97%产率)。
LC/MS:(MH)+=767
实施例19
制备Nα-Fmoc-Ala-Ala-Asn-PABC-阿霉素[19]
将在干燥DMF(5mL)中的Nα-Fmoc-Ala-Ala-Asn-PABC-PNP[18](383mg,0.5毫摩尔)、阿霉素盐酸盐(348mg,0.6毫摩尔)、HOB T(202mg,1.5毫摩尔)以及DIEA(325mg,2.5毫摩尔)的反应溶液在暗处搅拌过夜。在冰-水温度下向上述溶液中缓慢添加5%柠檬酸(100mL),并且搅拌1小时。通过过滤收集沉淀物,用5%柠檬酸随后用去盐水冲洗。将固体再悬浮于NaHCO3水溶液中并且搅拌30分钟。收集固体,用NaHCO3溶液随后用去盐水充分冲洗。将固体再悬浮于异丙醇,10%甲醇/乙酸乙酯中,研磨,过滤并且在真空中干燥,得到暗红色产物(550mg,94%产率)
LC/MS:MH)+=1171
实施例20
制备Ala-Ala-Asn-PABC-阿霉素[20]
将在20%吗啉/NMP(5mL)中的Nα-Fmoc-Ala-Ala-Asn-PABC-阿霉素[19](500mg,0.42毫摩尔)的溶液搅拌1小时。向上述反应溶液中缓慢添加乙醚(50mL)并且将所得悬浮液搅拌30分钟。倒出乙醚(重复三次)。将残余物在乙酸乙酯,20%甲醇/乙酸乙酯中研磨。通过过滤收集固体并且在真空中干燥。将固体再悬浮于水中,超声处理,过滤并且在真空中用KOH干燥,得到暗红色产物(320mg,80%产率)。
LC/MC:(MH)+=949
将用于制备实施例8、12和13的类似方法用于由化合物[20]来制备实施例21、22和23。
实施例21
Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-阿霉素[21]
LC/MS:(MH)+=1049
实施例22
Nα-[-(2-酰胺-2-氧代乙氧基)乙酸]-Ala-Ala-Asn-PABC-阿霉素[22]
LC/MS:(MH)+=1065
实施例23
Nα-[-((2-酰胺-2-氧代乙氧基)(甲基)氨基)乙酸]-Ala-Ala-Asn-PABC-阿霉素[23]
LC/MS:(MH)+=1078
实施例24
制备Nα-Fmoc-Asn-PABC-喜树碱[24]
在冰水温度下,在搅拌下向在干燥CH2Cl2(20mL)中的喜树碱(348mg,1毫摩尔)和DAMP(366mg,3毫摩尔)的悬浮液中添加三光气(100mg,0.33毫摩尔)。20分钟后,将在干燥CH2Cl2(10mL)中的Nα-Fmoc-Asn-PAB-OH[2](459mg,1毫摩尔)的悬浮液添加到上述反应混合物中并且在室温下搅拌过夜。向所得反应混合物中添加对-硝基苯氯甲酸酯(100mg,0.5毫摩尔),然后添加另外量的DAMP(60mg,0.5毫摩尔),并且将反应混合物再搅拌4小时。浓缩后,用25%CH2Cl2/乙酸乙酯研磨所得残余物,过滤,用25%CH2Cl2/乙酸乙酯冲洗并且在真空中干燥。将固体悬浮于NaHCO3水溶液中,超声处理,过滤,用NaHCO3水溶液,随后用5%柠檬酸、去盐水充分冲洗,并且在真空中用KOH干燥,得到灰黄色产物(763mg,91%产率)。
LC/MS:(MH)+=834
实施例25
制备Asn-PABC-喜树碱[25]
将在2%DBU/CH2Cl2(15mL)中的Nα-Fmoc-Asn-PABC-喜树碱[24](500mg,0.6毫摩尔)的溶液搅拌20分钟。将乙醚(80mL)缓慢添加到上述反应溶液中并且搅拌30分钟。研磨沉淀物,通过过滤收集并且用乙醚充分冲洗。将固体在20%CH2Cl2/乙酸乙酯中再研磨,过滤并干燥。将固体再悬浮于NaHCO3水溶液中,研磨,过滤,用去盐水冲洗并且在真空中用KOH干燥,得到灰黄色产物(295mg,80%产率)。
LC/MS:(MH)+=612
实施例26
制备Nα-Fmoc-Ala-Ala-Asn-PABC-喜树碱[26]
将在NMP(5mL)中的H2N-Asn-PABC-喜树碱[25](244mg,0.4毫摩尔)、Nα-Ala-Ala[1](190mg,0.5毫摩尔)、PyBop(260mg,0.5毫摩尔)以及DIEA(325mg,2.5毫摩尔)的溶液搅拌1小时。向上述反应溶液中缓慢添加5%柠檬酸(50mL)并且将所得混合物搅拌30分钟。通过过滤收集沉淀物,用5%柠檬酸随后用去盐水冲洗,并且在真空中用KOH干燥。将固体在15%CH2Cl2/乙酸乙酯中研磨,超声处理,过滤并且干燥,得到灰色产物(274mg,70%产率)。
LC/MS:(MH)+=976
实施例27
制备Ala-Ala-Asn-PABC-喜树碱[27]
将在2%DBU/CH2Cl2(10mL)中的Nα-Fmoc-Ala-Ala-Asn-PABC-喜树碱[26](487mg,0.5毫摩尔)的溶液搅拌10分钟。向上述反应溶液中缓慢添加乙醚(50mL)并且搅拌30分钟。收集沉淀物,用乙醚冲洗。将固体悬浮于20%CH2Cl2/乙酸乙酯中,研磨,过滤,用乙酸乙酯冲洗并且干燥,得到灰色产物(290mg,77%产率)。
LC/MS:(MH)+=754
将用于制备实施例8、12和13的类似方法用于由化合物[27]来制备实施例28、29和30。
实施例28
Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-喜树碱[28]
LC/MS:(MH)+=854
实施例29
Nα-[-(2-酰胺-2-氧代乙氧基)乙酸]-Ala-Ala-Asn-PABC-喜树碱[29]
LC/MS:(MH)+=870
实施例30
Nα-[-((2-酰胺-2-氧代乙氧基)(甲基)氨基)乙酸]-Ala-Ala-Asn-PABC-喜树碱[30]
LC/MS:(MH)+=883
实施例31
制备N1-Ala-Ala-Asn-PABC-丝裂霉素、N4-Ala-Ala-Asn-PABC-阿霉素-琥珀酰胺[31]
将在NMP(2mL)中的Nα-琥珀酰胺酸-Ala-Ala-Asn-PABC-丝裂霉素[8](85mg,0.1毫摩尔)、Ala-Ala-Asn-PABC-阿霉素[20](104mg,0.11毫摩尔)、PyBop(62mg,0.12毫摩尔)以及DIEA(78mg,0.6毫摩尔)的溶液搅拌1小时。添加乙醚(20mL),并且将所得混合物搅拌并超声处理。倒出乙醚(重复两次),并且将残余物在甲醇中研磨,过滤并且干燥,得到暗色产物(100mg,57%产率)
LC/MS:(MH)+=1770
实施例32
制备N1-Ala-Ala-Asn-PABC-丝裂霉素、N5-Ala-Ala-Asn-PABC-阿霉素-双(Oα)-乙酰胺[32]
将用于制备实施例31的类似方法用于由化合物[20]和化合物[12]来制备实施例32。
LC/MS:(MH)+=1786
实施例33
制备N1-Ala-Ala-Asn-PABC-丝裂霉素、N5-Ala-Ala-Asn-PABC-阿霉素-双(Nα-甲基)-乙酰胺[33]
将用于制备实施例31的类似方法用于由化合物[20]和化合物[13]来制备实施例33。
LC/MS:(MH)+=1799
实施例34
制备N1-Ala-Ala-Asn-PABC-丝裂霉素、N4-Ala-Ala-Asn-PABC-喜树碱-琥珀酰胺[34]
将用于制备实施例31的类似方法用于由化合物[27]和化合物[8]来制备实施例34。
LC/MS:(MH)+=1575
实施例35
制备N1-Ala-Ala-Asn-PABC-丝裂霉素、N5-Ala-Ala-Asn-PABC-喜树碱-双(Oα)-乙酰胺[35]
将用于制备实施例31的类似方法用于由化合物[27]和化合物[12]来制备实施例34。
LC/MS:(MH)+=1591
实施例36
制备N1-Ala-Ala-Asn-PABC-丝裂霉素、N5-Ala-Ala-Asn-PABC-喜树碱-双(Nα-甲基)-乙酰胺[36]
将用于制备实施例31的类似方法用于由化合物[27]和化合物[13]来制备实施例34。
LC/MS:(MH)+=1604
在实施例中使用的缩写:
DCC=二环己基碳二亚胺
DCM=二氯甲烷
DCU=二环己基脲
DIEA=二异丙基乙胺
DMA=二甲基乙酰胺
DMEM=Dulbecco改良的Eagle培养基
DMF=N,N-二甲基甲酰胺
DMSO=二甲基亚砜
EDC=1-乙基-3-(3-二甲基氨基丙基)碳二亚胺
EDTA=N,N',N",N"',N""-乙二胺四乙酸
FBS=胎牛血清
Fmon=芴基甲氧基氨基甲酰基
HATU=1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶-3-氧化物六氟磷酸盐
HOBT=N-羟基苯并三唑
HPLC=高压液相色谱
IR=红外光谱
LC/MS=液相色谱/质谱
MS=质谱
MTD=最大耐受剂量
NMP=N-甲基吡咯烷酮
NMR=核磁共振
PABC=对-氨基苄基氨基甲酰基
PBS=磷酸盐缓冲盐水
Py=吡啶
QW=每周
TCA=三氯乙酸
Tr=三苯甲基,三苯基甲基
UV/VIS=紫外/可见光谱
- 还没有人留言评论。精彩留言会获得点赞!