通过共沉淀制备亚微米级别的无定形固体分散体的方法与流程
1.发明领域
本发明涉及通过在装置中进行溶剂控制的共沉淀来制备无定形固体分散体的方法,所述装置促进所定义的反应室或微通道内的分子接触/相互作用,下文称作微反应技术(mrt)。特别地,本发明涉及获得具有亚微米级别的粒径的、颗粒形式的药学活性化合物(api)的无定形固体分散体的方法,通过所述方法获得的无定形固体分散体和它们的用途。所述方法可以应用于药学领域,特别是活性药学成分、中间药物产品或药物产品的加工。本文所述的方法与连续制备相容,并且允许在单一步骤中进行固体分散体的合成和颗粒工程学。此外,按照本发明的方法制备的无定形颗粒在粒径和密度方面表现出有利特征。
2.发明背景
多达90%的开发中的活性药学物质是水溶性很差的,通常导致低生物利用度。为了克服这点并且促进新化学实体成功转化为药品,已开发了不同的工程学和配制方法:颗粒设计和大小减小技术、自乳化药物递送系统、环糊精复合物、无定形固体分散体、盐形式和共结晶形式(cocrystalform)。在不同选择中,稳定的无定形固体分散体的使用正成为越来越受欢迎的平台,大量药物到达市场。无定形固体分散体包含至少两种组分,一般为稳定剂(例如聚合物)和药物。无定形固体分散体与其他配制策略相比的独特优势是,一旦药物开始在吸收部位溶解,就获得过饱和状态,即药物的浓度达到远远高于其固有溶解度的值。可以通过改善溶出动力学(可应用于iia类化合物,根据可展性分类系统(developabilityclassificationsystem),即dcs,或生物制药学分类系统(biopharmaceuticsclassificationsystem),即bcs)和/或通过增加活性化合物在溶液中的最大浓度(可应用于dcsiib类化合物)实现生物利用度的增强。在溶出动力学是实现生物利用度的关键的情况下,无定形固体分散体的性质,即粒径,可以在药物产品的溶出曲线方面起重要作用。
虽然文献中报道了用于制备固体分散体的各种方法(例如喷雾干燥、冷冻干燥、热熔挤出),但是现有技术缺少能够同时将粒径控制在亚微米级别和保持固体分散颗粒的无定形性质的技术。喷雾干燥的颗粒通常是中空的,具有低密度和2至120微米的粒径,相反来自热熔挤出的挤出物是致密的,具有非常粗糙的颗粒或小球,并且需要额外的下游加工(例如研磨)以获得精细材料。
本发明的目的之一是提供可选的共沉淀方法,其使用微反应或微流化以促进包含活性成分、诸如稳定剂的赋形剂以及溶剂/抗溶剂体系的流体(stream)之间的分子接触或相互作用,并且获得具有高密度的亚微米级别的无定形固体分散体。
在无机化合物的应用领域中,filipacastro等人(phdthesis,processintensificationfortheproductionofhydroxyapatitenanoparticles,univ.minho,2013)的微反应技术用来改善羟基磷灰石纳米颗粒的生产和特征。当与传统方法如搅拌容器相比时,filipacastro等人观察到优势,这是由于表面积比容积比例的增加增强热和质量转移。
在药物化合物的应用领域中,现有技术包括在单独药物的颗粒的加工和/或结晶材料的加工中的相似方法的一些实例。us8367004b2公开了制备具有亚微米级别的粒径的晶体或多晶型物的方法。hanyali等人(iranianjournalofpharmaceuticalresearch,13(3),2014,785-795)描述了制备布地奈德的纳米晶体的自底向上技术。hongzhao等人(ind.eng.chem.res.,46(24),2007,8229–8235)描述了一种制备亚微米区域的活性药物成分的单独药物结晶颗粒的方法。中国专利cn201337903ya还公开了制备无定形单独药物颗粒产品头孢呋辛酯的一种新方式的细节。虽然上述参考文献提供微流化或微反应的深刻见解及其益处,令人惊讶的是现有技术中从未评价无定形固体分散体的共沉淀。
术语“微反应”是指包括微反应器、微混合器、微通道或微流体领域内包含的任何其他组分内的物理和/或化学反应的技术。
术语“微流化”是指微流体反应技术(mrt),其涵盖诸如装置的硬件和方法。mrt可以用来通过最小化单相和多相反应物流体之间的扩散限制来制备纳米颗粒和/或加快化学反应速度。该技术包括通过固定几何体高剪切、连续流体加工,其提供中观和微观混合级别的剧烈和均匀混合并产生纳米级别的漩涡和产物。
术语“无定形固体分散体”定义为至少一种药物在基质中的分散体,所述分散体呈无定形状态。所述基质可以包含聚合物、表面活性剂或其混合物。在本发明的范围中,这个术语还用来描述共沉淀物,其呈包含活性成分和所述基质的无定形纳米颗粒的形式。
在药物无定形固体分散体的情况下,成分、溶剂和/或抗溶剂体系、每种组分各自的浓度以及混合条件的选择对于所有构成部分的同时沉淀或共沉淀至关重要,所述同时沉淀或共沉淀的方式使沉淀颗粒的组成对应于预期的制剂。本文公开的方法不仅解决了制备药物无定形固体分散体的挑战,而且还解决了将这样的颗粒系统控制在亚微米级别。因此,其适合解决溶出度和/或溶解度受限的药物化合物,通常被bcs命名为ii类化合物。
在通过溶剂控制的共沉淀制备亚微米颗粒的领域中,现有技术还包括许多有意义的实例。us7037528b2公开了一种方法,其中利用酸化冷水作为抗溶剂和肠溶聚合物作为分散剂,通过溶剂控制的沉淀获得共沉淀的颗粒。共沉淀步骤之后,该专利描述了高能步骤的执行,以便获得0.4μm至2.0μm的粒径。但是,通过这种方法产生共沉淀的颗粒不允许控制材料的固体状态,其可以是无定形的、结晶的或半结晶的。此外,共沉淀步骤之后第三步骤的使用表明通过使颗粒处于高能条件来稳定粒径。us7037528b2的另一局限是指制剂中表面活性剂的强制使用。本发明解决所有这些局限。
wo2013105894a1描述了一种通过混合两个流体和经喷嘴将混合物喷雾以及干燥来制备无定形混合纳米颗粒的制备方法。这种方法在一个流体中使用超临界流体以沉淀材料,然后雾化,并且收集喷雾干燥的颗粒。虽然这种方法适合制备具有亚微米级别的粒径的无定形固体分散体,但是其受到化合物在超临界流体(通常是二氧化碳)中的溶解度的限制,并且增加用高压和高温气体加工进料的挑战,引入的超临界流体对于商业制备是严重障碍。
因此,本发明的发明人已意识到需要提供制备无定形固体分散体、特别是亚微米级别的无定形固体分散体的方法。这通过本发明的方法得以实现,其能够在单一制备步骤中制备具有低至至少50nm的稳定大小的药物无定形固体分散颗粒。所述方法使用微反应或微流化以促进包含活性成分的流体之间的分子接触或相互作用以获得具有高密度的亚微米级别的无定形固体分散体。而且,所述技术适用于连续加工,并且可容易地扩展至商业规模。此外,将成分的溶解度限制最小化,因为它们可以溶解于溶剂体系和/或抗溶剂体系。
3.发明概述
根据本发明的一方面,提供制备颗粒形式的无定形固体分散体的方法,所述方法包括:(i)制备包含至少一种药学活性化合物的溶液和包含至少一种稳定剂的溶液,其中利用第一溶剂制备每一种溶液,以及(ii)通过微流化或微反应将所述溶液与包含至少一种抗溶剂的第二溶剂混合以通过共沉淀获得无定形颗粒的悬浮液。
优选地,在与所述第二溶剂混合之前,将所述包含至少一种药学活性化合物的溶液和所述包含至少一种稳定剂的溶液合并以形成第一流体,所述第二溶剂可以是所述药学活性成分和所述稳定剂的抗溶剂。优选地,将所述包含所述稳定剂的溶液与所述第二溶剂合并以形成第二流体,所述第二流体包含所述药学活性化合物的抗溶剂。
根据本发明的另一方面,提供制备颗粒形式的无定形固体分散体的方法,所述方法包括:(i)利用第一溶剂制备包含至少一种药学活性化合物的溶液和利用第二溶剂制备包含至少一种稳定剂的溶液;其中所述第二溶剂是所述药学活性化合物的抗溶剂;以及(ii)通过微流化或微反应混合所述溶液以通过共沉淀获得无定形颗粒的悬浮液。
所述方法优选包括分离步骤以分离粉末形式的无定形颗粒。
根据本发明的另一方面,提供通过本发明的方法获得的颗粒无定形固体分散体,所述分散体包含5%至95%(w/w)的所述药学活性化合物和95%至5%(w/w)的所述稳定剂。所述稳定剂优选是至少一种表面活性剂和/或聚合物。
根据本发明的另一方面,提供包含如本文所述的颗粒无定形固体分散体的药物组合物。
根据本发明的另一方面,提供包含颗粒无定形固体分散体的药物组合物,其用作药物。
根据本发明的另一方面,提供颗粒无定形固体分散体,其用于增加药学活性化合物的生物利用度。
当考虑本发明的以下详细描述和附图时会更容易理解本发明的前述和其他特点和优势。
4.附图说明
这些图说明本发明的一些实施方案的某些方面,并且不应当用来限制或定义本发明。
图1示出本发明方法的示意图。
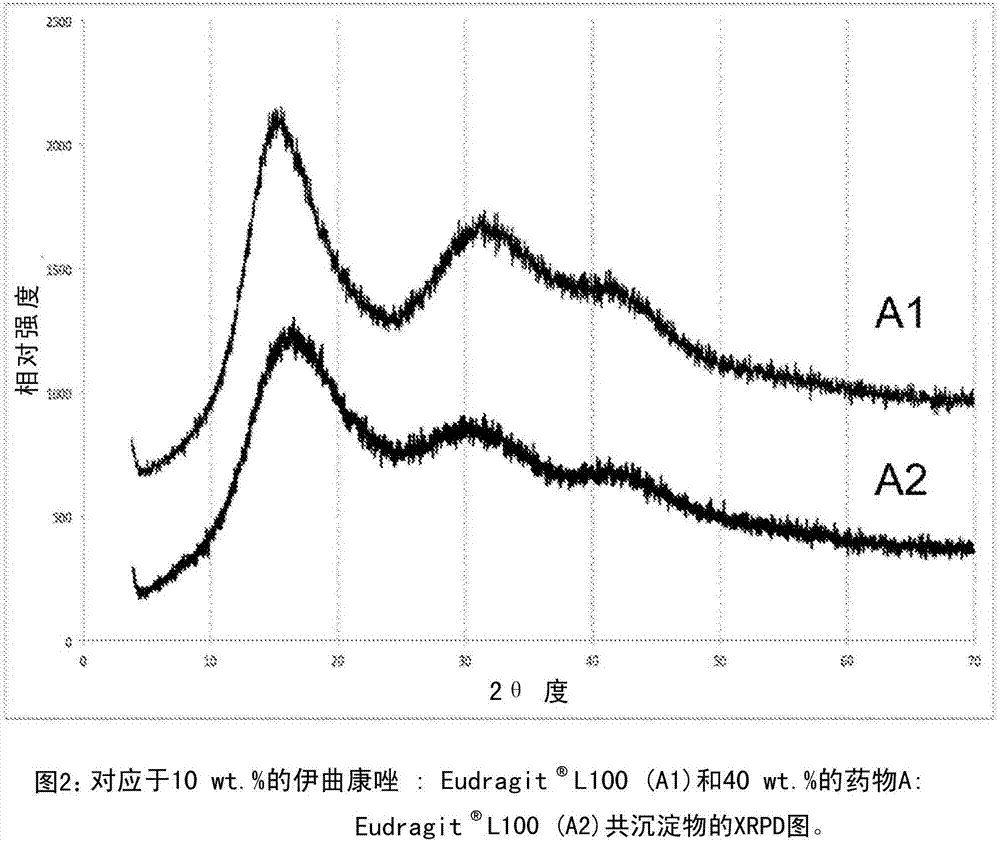
图2示出对应于10wt.%和40wt.%的药物负荷伊曲康唑:
图3示出对应于通过过滤加上在托盘烘箱中干燥而分离的(b1)以及通过喷雾干燥而分离的(b2)10wt.%的桂利嗪:
图4示出对应于通过喷雾干燥而分离的10wt.%的桂利嗪:
图5示出对应于10wt.%和40wt.%的药物负荷尼洛替尼:
图6示出40wt.%的尼洛替尼:
图7示出对应于共沉淀之后(a)和通过喷雾干燥而分离之后(b)的20wt.%的卡马西平:
图8示出对应于20wt.%的卡马西平:
图9示出对应于20wt.%的卡马西平:
图10示出在本发明的范围下制备的材料(nanoamorphous–a)和通过喷雾干燥制备的材料(microamorphous-b)的粉末体外溶出曲线。
图11示出具有20wt.%的卡马西平:
5.发明详述
本发明提供制备颗粒形式的无定形固体分散体的方法,所述无定形固体分散体包含至少一种药学活性化合物和至少一种稳定剂,具有亚微米级别的粒径。
如图1所示,所述制备方法可以分为3个阶段:
阶段(i)–选择两种溶剂(所述第一溶剂和所述第二溶剂)。选择所述溶剂,从而所关注的活性药学化合物在一种溶剂(下文称作“溶剂”)中部分可溶,并且在另一种溶剂(下文称作“抗溶剂”)中基本上不溶。在一优选方面,所述药学活性化合物和所述稳定剂(即,聚合物和/或表面活性剂)溶解或部分溶解于所述第一溶剂中,并且所述第二溶剂包含所述药学活性化合物和所述稳定剂的抗溶剂。在一优选方面,所述药学活性化合物溶解或部分溶解于所述第一溶剂中,并且所述第二溶剂是所述药学活性化合物的抗溶剂。在一优选方面,所述稳定剂(即,表面活性剂、聚合物)溶解或部分溶解于所述第二溶剂中,所述第二溶剂是所述药学活性化合物的抗溶剂。术语“抗溶剂”在本文中用来描述在其中所述物质/化合物表现出非常低的溶解度的溶剂。当将多于一种的物质/化合物与常用抗溶剂混合时,所述物质在所述抗溶剂内沉淀而不是溶解,优选形成由所述不同物质制成的复合颗粒。
优选地,术语“抗溶剂”在本文中用来描述一种溶剂或溶剂的混合物,其中当与溶剂相比时,所述物质表现出非常低的溶解度。优选地,术语“抗溶剂”用来指在其中所述物质完全不溶的溶剂。
优选地,术语“溶剂”在本文中用来描述溶剂或溶剂的混合物,在其中所述物质表现出高达50体积/g溶质的比例的溶解度。术语“体积/g”是指毫升溶剂/克溶质的度量。
本领域技术人员能够选择可以用作特定化合物的溶剂和另一化合物的抗溶剂的溶剂。
优选地,术语“可溶的”表示需要10至30份溶剂以溶解1份溶质。
优选地,术语“非常低的溶解度”表示需要100至1000份溶剂以溶解1份溶质。
优选地,术语“基本上不溶的”表示需要1000至10000份溶剂以溶解1份溶质;而术语“不溶的”表示需要超过10000份溶剂以溶解1份溶质。
术语“溶剂”份和“溶质”份是指以毫升计的适当体积的溶剂/克溶质。
在阶段(i)中,制备第一流体(1),其包含能够在至少一种溶剂/抗溶剂体系中形成共沉淀物的一种或多种药学活性化合物以及一种或多种稳定剂(即,聚合物和/或表面活性剂)的溶液。优选地,单独地制备一种或多种药学活性化合物的溶液以及一种或多种稳定剂(即,聚合物和/或表面活性剂)的溶液,然后合并以形成所述第一流体。制备包含第二溶剂的第二流体(2),所述第二溶剂包含所述药学活性化合物和所述稳定剂的抗溶剂。
在一优选方面,将所述一种或多种稳定剂的溶液与所述第二溶剂合并以形成第二流体,或者可以将所述稳定剂直接添加至所述第二溶剂以形成第二流体。在这种情况下,所述第二溶剂可以充当所述稳定剂的溶剂,并且充当所述第一流体中存在的所述药学活性化合物的抗溶剂。
优选地,所述溶液中存在的所述药学活性化合物比所述稳定剂的比例为约95:5(%w/w)至约5:95(%w/w)。
阶段(ii)–将所述第一流体(1)和所述第二流体(2)在用于实现微流化或微反应的装置(3)中于受控条件下混合。用于实现微流化的装置优选是微型反应器或微流体反应技术(mrt)设备,或者促进所定义的反应室或微通道内的高效分子接触/相互作用的任何相似设备,以便通过两个流体中的物质的共沉淀形成无定形纳米颗粒的悬浮液(4)。优选地,所述反应室包括具有明确定义的直径和大小的一个或多个通道。优选地,所述通道的直径为约10微米至约400微米。更优选地,所述直径为约50微米至约200微米。一个或多个装置,例如微型反应器或mrt,可以串联或并联使用。优选地,所述装置中进行的过程是连续过程。
优选地,将所述溶液连续泵入所述反应室,在所述反应室中所述溶液被混合并被允许反应(连续流动反应)。
优选地,将所述第一流体和所述第二流体以不同的、单独控制的速度输送到一个或多个增压泵,从而基本上控制所述第一流体和所述第二流体之间的相互作用,然后将所述流体输送至所述用于微流化或微反应的装置(3)。优选地,通过使用至少一个增压泵来控制整体流速,即包含活性物质、赋形剂(稳定剂)、溶剂和抗溶剂的两个流体的流速。所述整体流速可以高达50kg/h。
优选地,可以使用蠕动泵来控制所述两个流体(即溶剂流和抗溶剂流)中至少一个的流速。所述两个流体的流速可以单独控制,因此每一个流体的流速可以为约0至50kg/h。
然后,用一个或多个增压泵将所述第一流体和所述第二流体以合并的流体的形式于升高的压力下加压并递送至所述装置,导致所述第一流体和所述第二流体的成分在所述装置内在纳米/微米级水平相互作用。优选地,过程压力是(但不限于)约345巴至约3500巴。
混合比例、过程压力和固体浓度的选择应当优化,以达到期望的粒径。优选地,溶剂比抗溶剂的混合比例是(但不限于)约1:2至1:50。优选地,溶剂混合物中的固体浓度是(但不限于)约1%至30%w/w。
优选地,共沉淀条件如溶剂/抗溶剂体系和混合条件决定所制备的固体的无定形性质、粒径和形态学。溶剂比抗溶剂的比例取决于所使用的物质和溶剂的特征,如溶剂的过饱和能力和物质的沉淀速度。优选地,抗溶剂比例是溶剂比例的2至30倍。
优选地,溶剂和抗溶剂体系的温度控制用来操纵过饱和能力和物质的沉淀速度。优选地,具有所述成分的溶剂和/或抗溶剂体系的温度是(但不限于)-10℃至50℃。
阶段(iii)–本发明的方法包括任选存在的分离步骤(5),以通过从所得的包含活性药物和稳定剂的固体分散体去除溶剂,分离粉末形式的无定形纳米颗粒(6)。可以通过本领域技术人员已知的任何合适技术去除溶剂。优选地,去除溶剂的步骤包括蒸馏、干燥、喷雾干燥、过滤或它们的任何组合。还可以在分离过程参数中控制所获得的无定形固体分散颗粒的形态学。例如,可以控制所述无定形固体分散颗粒的形态学,例如团聚水平、聚集体的孔隙率和粉末的堆密度,优选通过喷雾干燥过程中使用的雾化和/或在一定温度下的干燥过程去除最终产物中的过量残余溶剂来控制。
本发明的方法包括制备包含至少一种药学活性化合物的溶液和包含至少一种稳定剂的溶液。优选地,制备包含所述药学活性化合物和所述稳定剂的溶液。
所述药学活性化合物和所述稳定剂能够在至少一种溶剂和/或抗溶剂体系中形成无定形共沉淀物。然后通过微流化或微反应将所述一种或多种溶液与第二溶剂混合,以通过共沉淀获得无定形颗粒的悬浮液。所述第二溶剂包含所述药学活性化合物和/或所述稳定剂的至少一种抗溶剂。可以已知方式分离粉末形式的无定形颗粒。所述无定形颗粒是具有亚微米级别的粒径的纳米颗粒。在本发明的上下文中,术语“亚微米级别”是指几nm至少于1mm的粒径。
所述无定形纳米颗粒的粒径可以是约50nm至约10μm;优选约50nm至约1μm;并且更优选约50nm至约500nm。
在一优选方面,所述方法包括:(i)利用第一溶剂制备包含至少一种药学活性化合物的溶液和利用第二溶剂制备包含至少一种稳定剂的溶液;其中所述第二溶剂是所述药学活性化合物的抗溶剂;以及(ii)通过微流化或微反应混合所述溶液以通过共沉淀获得无定形颗粒的悬浮液。
在一优选方面,所述方法包括:(i)利用第一溶剂制备包含至少一种药学活性化合物和至少一种稳定剂的溶液;以及(ii)通过微流化或微反应将所述溶液与第二溶剂混合以通过共沉淀获得无定形颗粒的悬浮液,所述第二溶剂包含所述药学活性化合物和所述稳定剂的至少一种抗溶剂。
在一优选方面,所述方法包括:(i)制备包含至少一种药学活性化合物的溶液和包含至少一种稳定剂的溶液,其中利用第一溶剂制备每一种溶液;以及(ii)通过微流化或微反应将所述溶液与第二溶剂混合以通过共沉淀获得无定形颗粒的悬浮液,所述第二溶剂包含所述药学活性化合物和所述稳定剂的至少一种抗溶剂。优选地,在与所述第二溶剂混合之前,将所述溶液合并以形成单一流体。
优选地,将所述包含至少一种药学活性化合物的溶液和所述包含至少一种稳定剂的溶液合并以形成第一流体,然后通过微流化或微反应与所述第二溶剂混合。优选地,所述第二溶剂是所述药学活性成分和所述稳定剂的抗溶剂。
优选地,将所述包含所述稳定剂的溶液与所述第二溶剂合并以形成第二流体。所述第二流体可以包含所述药学活性化合物的抗溶剂。在这种情况下,所述包含所述药学活性化合物的溶液形成第一流体。
本发明的方法中使用的稳定剂可以包含至少一种聚合物和/或至少一种表面活性剂。所述聚合物和/或表面活性剂可以所述固体分散体的约0.001%至90%(w/w)的量存在。所述表面活性剂优选选自:阴离子表面活性剂、阳离子表面活性剂、非离子表面活性剂和它们的组合。
优选地,将所述包含所述药学活性化合物和所述稳定剂的第一流体与所述包含所述药学活性化合物和所述稳定剂的抗溶剂的第二流体在足以引起所述流体中的成分的相互作用的压力下合并;并且递送至所述反应室中的所述一个或多个通道,从而所述流体中的所述成分在微米和/或纳米级别发生反应以通过共沉淀形成无定形纳米颗粒的悬浮液。
优选地,其中将所述包含所述药学活性化合物的第一流体与所述包含所述稳定剂以及所述药学活性化合物的抗溶剂的第二流体在足以引起所述流体中的成分的相互作用的压力下合并,并且递送至所述反应室中的所述一个或多个通道,从而所述流体中的所述成分在微米和/或纳米级别发生反应以通过共沉淀形成无定形纳米颗粒的悬浮液。
本发明的方法可以进一步包括在所述mrt或微型反应器内相互作用之后使合并的流体冷却或急冷(quenching)的步骤。
本发明还涉及颗粒无定形固体分散体,其包含5%至95%(w/w)的所述药学活性组分和95%至5%(w/w)的所述稳定剂,所述稳定剂优选为表面活性剂和/或聚合物。所述颗粒的粒径可以是约50nm至约10μm,优选约50nm至约1μm,更优选50nm至约500nm。
优选地,所述固体分散体的颗粒具有约0.1g/ml至1.0g/ml的堆密度。
用于本发明的方法的有机化合物是其溶解度从一种溶剂至另一种溶剂减少的任何有机化学实体。这种有机化合物优选是一种或多种药学活性化合物。优选的药学活性化合物的实例可以包括(但不限于)难溶性活性化合物,稳定性差的热不稳定化合物,或者需要小粒径和高密度的药物产品。
在一优选方面,“低溶解度”、“难溶性”和“水难溶性”化合物的定义对应于生物药剂学分类系统(bcs)的定义。根据bcs,关于溶解度(根据美国药典)和肠通透性,可以将化合物分为4类。i类化合物具有高通透性和高溶解度,ii类化合物具有高通透性和低溶解度,iii类化合物的特征在于低通透性和高溶解度,而iv类化合物具有低通透性和低溶解度。难溶性化合物对应于ii类和iv类。
难溶性化合物的实例包括、但不限于:抗真菌剂如依他康唑(intraconazole)或相关药物,如氟康唑(fluoconazole)、特康唑、酮康唑和沙康唑;抗感染药物,如灰黄霉素和相关化合物(例如griseoverdin);疟疾药物(例如阿托伐醌);蛋白激酶抑制剂如阿法替尼(afatinib)、阿昔替尼、波舒替尼、西妥昔单抗、克里唑蒂尼(crizotinib)、达沙替尼、厄洛替尼(erlotinib)、fostamatinib、吉非替尼、依鲁替尼(ibrutinib)、伊马替尼、zemurasenib、拉帕替尼、乐伐替尼(lenvatinib)、莫立替尼或尼洛替尼;免疫系统调节剂(例如环胞素);心血管药物(例如地高辛和螺内酯);布洛芬;固醇或类固醇;来自包含以下的组的药物:达那唑、阿昔洛韦、氨苯砜、茚地那韦、硝苯地平、呋喃妥因(nitrofurantion)、苯妥英(phentytoin)、利托那韦、沙奎那韦、磺胺甲噁唑、丙戊酸、甲氧苄啶、乙酰唑胺、硫唑嘌呤、碘番酸、萘啶酸、奈韦拉平、吡喹酮、利福平、阿苯达唑、阿米替林、蒿甲醚、本芴醇、氯丙嗪、环丙沙星、氯法齐明、依法韦仑、洛匹那韦(iopinavir)、叶酸、格列本脲、氯哌啶醇、伊维菌素、甲苯达唑、氯硝柳胺、噻嘧啶、乙胺嘧啶、视黄醇维生素、磺胺嘧啶、柳氮磺嘧啶、三氯苯达唑和桂利嗪。
优选难溶性化合物的组的详细列表包括、但不限于单独的以下药剂或其组合:ace抑制剂组的活性剂或生物活性化合物、腺垂体激素、肾上腺素能神经元阻断剂、肾上腺皮质类固醇,肾上腺皮质类固醇生物合成的抑制剂、α-肾上腺素能激动剂、α-肾上腺素能拮抗剂、选择性α2-肾上腺素能激动剂、镇痛剂、退热剂和抗炎剂、雄激素、麻醉剂、抗成瘾剂(antiaddictiveagent)、抗雄激素剂、抗心律失常剂、平喘剂、抗胆碱剂、抗胆碱酯酶剂、抗凝剂、抗糖尿病剂、止泻剂、抗利尿剂、止吐和促动力剂、抗癫痫剂、抗雌激素剂、抗真菌剂、抗高血压剂、抗微生物剂、抗偏头痛剂、抗毒蕈碱剂、抗肿瘤剂、抗寄生虫剂、抗帕金森剂、抗血小板剂、抗孕素、抗甲状腺剂、镇咳剂、抗病毒剂、抗抑郁剂、氮杂螺旋癸二酮类、巴比妥酸盐、苯二氮
优选地,所述药学活性化合物为酪氨酸激酶抑制剂。例如,其可以选自:阿昔替尼、克里唑蒂尼、达沙替尼、厄洛替尼、吉非替尼、伊马替尼、拉帕替尼、尼洛替尼、帕唑帕尼、瑞戈非尼、鲁索替尼(ruxolitinib)、索拉非尼、舒尼替尼、凡他尼布(vandetanib)、威罗菲尼(vemurafenib)和它们的组合。
所述药学活性化合物的优选实例包括、但不限于伊曲康唑、桂利嗪、尼洛替尼、卡马西平或它们的组合。
优选地,所述药学活性化合物是尼洛替尼。
所述药学活性化合物可以所述分散体的约0.1%至约90%(w/w)的量存在。
根据本发明,所述方法中使用的溶剂优选是一种溶剂或溶剂的混合物,在其中,一种或多种有机化合物(优选所关注的药学活性化合物)至少是部分可溶的。优选地,所述溶剂或溶剂的混合物还是这样的溶剂或溶剂的混合物,在其中,诸如稳定剂(聚合物/表面活性剂)的赋形剂至少是部分可溶的。所述溶剂可以具有一种或多种表面改性剂(表面活性剂),其优选是阴离子表面活性剂、阳离子表面活性剂或非离子表面活性剂。表面活性剂是降低两种液体之间或者液体和固体之间的表面张力(或界面张力)的化合物。表面活性剂还可以充当去污剂、湿润剂、乳化剂、起泡剂和/或分散剂。
这类溶剂的实例包括、但不限于:水、丙酮、氯甲烷、二甲基甲酰胺、甲醇、乙醇、二甲基亚砜、甲基乙基酮、二甲基乙酰胺、乳酸、异丙醇、3-戊醇、正丙醇、甘油、丁二醇、乙二醇、丙二醇、二甲基异山梨醇酯(dimethylisosorbide)、四氢呋喃、1,4-二噁烷、聚乙二醇、聚乙二醇酯、聚乙二醇山梨聚糖、聚乙二醇单烷基醚、聚丙二醇、聚丙烯藻酸酯(polypropylenealginate)、丁二醇或它们的混合物。
根据本发明,抗溶剂可以与溶剂互溶或不互溶,并且存在的物质在混合时表现出低溶解度或完全不溶。优选的抗溶剂是(但不限于)水溶液,其可以具有混合于其中的一种或多种表面改性剂如阴离子表面活性剂、阳离子表面活性剂或非离子表面活性剂。优选地,所述水溶液包含去离子水。
优选地,至少一种表面活性剂还可以用作稳定剂。所述表面活性剂是(但不限于)阴离子表面活性剂、阳离子表面活性剂、非离子表面活性剂或它们的组合。
合适的阴离子表面活性剂包括、但不限于月桂酸钾、月桂基硫酸钠、十二烷基硫酸钠、烷基聚氧乙烯硫酸盐、藻酸钠、二辛基磺基琥珀酸钠、磷脂酰胆碱、磷脂酰甘油、磷脂酰肌苷、磷脂酰丝氨酸、磷脂酸和它们的盐、羧甲基纤维素钠、胆酸和其他胆汁酸(例如,胆酸、脱氧胆酸、甘胆酸、牛磺胆酸、甘氨脱氧胆酸)和它们的盐(例如,脱氧胆酸钠等),或者其组合。
合适的阳离子表面活性剂包括、但不限于季铵化合物,如苯扎氯铵、溴化十六烷基三甲铵、月桂基二甲基苄基氯化铵、酰基肉碱盐酸盐或烷基吡啶卤化物。
合适的非离子表面活性剂包括、但不限于聚氧乙烯脂肪醇醚、聚氧乙烯脱水山梨糖醇脂肪酸酯、聚氧乙烯脂肪酸酯、脱水山梨糖醇酯、甘油单硬脂酸酯、聚乙二醇、聚丙二醇、鲸蜡醇、十六醇十八醇混合物、十八烷醇、芳基烷基聚醚醇、聚氧乙烯-聚氧丙烯共聚物(泊洛沙姆(poloxomers))、泊洛沙胺(polaxamines)、甲基纤维素、羟基纤维素、羟丙基纤维素、羟丙基甲基纤维素、非晶态纤维素、聚乙烯醇、甘油酯和聚乙烯吡咯烷酮或者它们的组合。
优选地,可以将ph调节剂添加至抗溶剂溶液。ph调节剂的实例包括、但不限于氢氧化钠、盐酸、氨丁三醇缓冲液或柠檬酸盐、乙酸盐、乳酸盐、葡甲胺等。
优选地,至少一种聚合物也可以用于所述无定形形式的稳定。适合用于本发明的制剂的聚合物包括但不限于纤维素酯、纤维素醚、聚亚烷基氧化物、聚丙烯酸酯、聚甲基丙烯酸酯、聚丙烯酰胺、聚乙烯醇、乙酸乙烯酯聚合物、寡糖、多糖、羟丙基纤维素、聚乙烯吡咯烷酮、羟基烷基纤维素、羟基烷基烷基纤维素、羟丙基甲基纤维素、邻苯二甲酸纤维素、琥珀酸纤维素、邻苯二甲酸醋酸纤维素、邻苯二甲酸羟丙基甲基纤维素、琥珀酸醋酸羟丙基甲基纤维素、聚环氧乙烷、聚环氧丙烷、环氧乙烷和环氧丙烷的共聚物、甲基丙烯酸/丙烯酸乙酯共聚物、甲基丙烯酸/甲基丙烯酸甲酯共聚物、琥珀酸羟丙基甲基纤维素、甲基丙烯酸丁酯/2-二甲氨基乙基甲基丙烯酸酯共聚物、聚(丙烯酸羟烷基酯)、聚(甲基丙烯酸羟烷基酯)、明胶、明胶、乙酸乙烯酯和巴豆酸的共聚物、部分水解的聚乙酸乙烯酯、角叉菜胶、半乳甘露聚糖、高粘度树胶或黄原胶,或者它们的组合。
除了活性化合物和稳定剂,混合物还可以包含用于提高制剂性能的其他物质。这些物质可以包含(但不限于)具有增塑效果的化合物或者具有改善活性物质的溶出曲线的性质的化合物。
可以将通过本发明的方法获得的颗粒配制为药物组合物。用于给药通过本文所述的方法合成的无定形固体分散体的药物形式的实例可以包括固体剂型,如片剂、胶囊剂、颗粒剂、小球或散剂。所获得的组合物可以具有增强的性能,包括(但不限于)过饱和、溶出度改善、控制释放或掩味。
本发明一般涉及制备颗粒形式的无定形固体分散体的方法,所述方法包括:制备包含能够在溶剂中形成无定形共沉淀物的至少一种活性药物成分和稳定剂的溶液;通过微型反应器将所述溶液与至少一种抗溶剂混合,通过共沉淀获得无定形纳米颗粒的悬浮液;任选地,所述方法可以包括分离步骤,以分离粉末形式的颗粒。所述溶液和所述抗溶剂在所述微型反应器中的相互作用有效定义了纳米悬浮液的无定形性质。通过微流化和空化,促进了所述微型反应器内的混合。优选地,在高压下进行所述溶液和所述抗溶剂的混合。
与现有技术中已知的制备无定形固体分散体的常规方法相比,本发明的方法表现出几个优势。用于制备无定形固体分散体的常规方法,如喷雾干燥和热熔挤出,不适合制备在挥发性有机溶剂中具有低溶解度的化合物和/或具有高熔点的化合物。本发明的方法可以使用广泛的溶剂/抗溶剂体系,同时避免使用高加工温度。可以操纵过程和配制条件(如混合能量、溶剂/抗溶剂比例、温度、停留时间、组成和浓度)以获得期望的颗粒性质,如粉末密度、粒径、表面积和溶出度。通过本发明的方法获得的悬浮颗粒始终处于无定形固体状态。而且,通过本发明的方法制备的颗粒无定形固体分散体是亚微米级别的,其是稳定的并且具有高密度。
此外,通过本发明的方法获得的无定形固体分散体是稳定的颗粒形式,避免了随后的稳定步骤。
通过本发明的方法获得的纳米颗粒的粒径在亚微米级别,避免了随后的可以导致固体状态改变的高能加工,例如研磨、高剪切混合。
而且,在本发明中,可以进行所述颗粒的分离,同时保持粉末特征。还可以进行所述颗粒的分离以调整/改善粉末特征。另一优势是所述方法可以适应连续加工并且其容易放大。
本发明还涉及包含本发明的颗粒无定形固体分散体的药物组合物。
本发明还涉及包含本发明的颗粒无定形固体分散体的药物组合物,其用作药物。
本发明还涉及颗粒无定形固体分散体,其用于增加药学活性化合物的生物利用度。
合适的实施例(其仅表示建议实施本发明的方法且不用来限制本发明的范围)如下:
实施例1
制备伊曲康唑(bcs/dcsiib类,tm/tg~1.32,logp4.4)和1:1甲基丙烯酸–甲基丙烯酸甲酯共聚物(
利用包括具有75μm直径的反应通道的室和随后的具有200μm直径的反应通道的室的purenanotm微流体反应技术(modelmrtcr5)制备共沉淀物。设置蠕动泵以维持溶剂和抗溶剂1:10的比例。设置增压泵以施加约20kpsi的压力。
共沉淀过程之后,利用真空,过滤所获得的悬浮液,并且将湿滤饼在托盘烘箱中于约70℃的温度下干燥~48h。在干燥过程结束时,通过x-射线粉末衍射(xrpd)分析粉末以进行固体状态分析,即评价结晶材料的潜在存在。
图2示出对应于10wt.%和40wt.%的药物负荷伊曲康唑:
两种制剂均表现出无定形形式的晕特征。在新鲜制备的产物中未观察到纯结晶伊曲康唑的xrpd谱的特征峰。这些结果表明,当使用本文所述的方法时,伊曲康唑的无定形化是成功的。此外,归功于还赋予稳定作用的聚合物的存在,能够制备高达40wt.%药物负荷的无定形制剂。
实施例2
制备桂利嗪(bcsii类,tm/tg~1.40,logp~5.77)和1:1甲基丙烯酸–甲基丙烯酸甲酯共聚物(
图3示出对应于通过过滤加上在托盘烘箱中干燥而分离的(b1)以及通过喷雾干燥而分离的(b2)10wt.%的桂利嗪:
实施例3
制备尼洛替尼(bcsiv类,tm/tg~1,28,clogp~4.8)和1:1甲基丙烯酸–甲基丙烯酸甲酯共聚物
图5示出对应于10wt.%和40wt.%的药物负荷尼洛替尼:
实施例4
还按照上述条件进行用40wt.%的尼洛替尼和60wt.%的
出于比较的目的,利用常规方法通过喷雾干燥制备相同的基于尼洛替尼的制剂——由于颗粒微米级别的,将制备的粉末在此后命名为microamorphous。实验条件与实施例2中提出的相似。
在它们的体外溶出曲线方面比较两种粉末。还评价结晶状态的溶出曲线。用37±0.5℃的恒温浴,利用uspii型装置(dis6000,copleyscientific,nottingham,uk)在900ml的ph6.5fassifbiorelevent介质(biorelevant,croydon,uk)中于100rpm的桨叶旋转下获得粉末溶出曲线。在非漏槽(non-sink)条件下用所研究的200mg尼洛替尼的靶药物负荷进行溶出实验。在各种时间点(15、30、60、120和240min)采集样品等分试样(15ml),不进行溶出介质置换。利用0.45μm滤器(具有0.45μm亲水聚丙烯(ghp)膜的
在分析体外溶出结果之前,进行利用量筒的密度测量,并且观察到nanoamorphous粉末的堆密度值和振实密度值是对于microamorphous粉末而获得的值的约2倍(即,对于堆密度,0.432g/ml对0.215g/ml,而对于振实密度,0.480g/ml对0.239g/ml)。
喷雾干燥藉由表现出低密度和差流动性的空心颗粒的制备被知晓。相反,在本发明的范围下制备的粉末是实心的/致密的,具有高堆密度和良好的流动性,对于下游加工(即压片、胶囊填充)是理想的。
图6示出40wt.%的尼洛替尼:
正如所预期的,nanoamorphous制剂和microamorphous制剂表现出比结晶的参比产物高的溶出度。
当比较nanoamorphous对microamorphous时,在15-分钟的时间点,观察到与后者相比,前者制剂达到显著较高的过饱和水平。在与无定形微固体分散体相比时,由于通过溶剂控制的沉淀过程制备的纳米颗粒的高表面积而引起的溶出度的增加有利于产生较高的过饱和水平。
实施例5
制备卡马西平(bcs/dcsiia类,tm/tg~1,4,logp~2,6)和1:1甲基丙烯酸–甲基丙烯酸甲酯共聚物(
为了评价通过本发明公开的方法制备的无定形纳米复合颗粒的体外性能,进行粉末溶出实验。
出于比较的目的,按照常规方法通过喷雾干燥制备具有20wt.%的卡马西平:
图10示出在本发明的范围下制备的材料(nanoamorphous-a)和通过喷雾干燥制备的材料(microamorphous-b)的粉末体外溶出曲线。在50-分钟时间点的虚线对应于ph-转变。
利用微量离心机ph-转变溶出方法获得粉末溶出曲线。在2ml微量离心管中于37℃温度水浴中进行实验。模拟胃相由0.9ml的0.01nhcl(ph=2)组成,而模拟肠相由0.9ml额外体积的fassifbiorelevent介质(ph=6.5)(biorelevant,croydon,uk)组成。利用ph试纸验证合并的ph值(ph6至6.5)。使用之前将两种介质脱气并预热至37℃。在非漏槽条件下用所研究的850ug卡马西平的靶药物负荷进行溶出实验,这分别对应于卡马西平在胃相和肠相中的平衡浓度的约5倍和2倍。在多个时间点(10、20、35、60、90、150和180min)采集样品等分试样,不进行溶出介质置换。ph转变发生在50-分钟时间点。用于采样的试管的准备由从水浴取出后者组成,并且利用himac微型离心机ct15re(hitachikokico,ltd)以13000rpm离心1分钟。然后,等分25ul的上清液,但是仅10ul用低体积插入管(150ul)在hplc小瓶中于甲醇中稀释15倍。将剩余的溶液涡旋几秒直至观察到沉淀物完全重新分散。将试管放回水浴直至下个时间点。利用具有光二极管阵列检测器(型号2996)的waters(型号2695)hplc系统进行溶解在介质中的卡马西平的量。使用的柱为
与以前的实施例相似,由于在本发明的范围下制备的无定形纳米颗粒的高表面积,与通过喷雾干燥制备的microamorphous相比,溶出度最大化。
为了理解在dcsiia类药物的吸收和生物利用度方面的协同效应(纳米+无定形),在小鼠中进行nanoamorphous制剂和microamorphous制剂的药代动力学研究。
成年cd1雌性小鼠(22至24g)购自charlesriver(barcelona,spain)。用标准实验室食物和水随意饲养动物。经当地动物伦理委员会批准并按照葡萄牙法律d.r.n°31/92,d.r.153i-a67/92和以下所有立法进行所有动物实验。在给药当天,在开始实验之前将动物禁食约6h。据认为这段时间足以排空小鼠的胃(labanim.2013,47(4),225-40)。通过口服管饲法用等量的每种制剂向小鼠给药以提供7.4mg/kg体重的卡马西平(n=3)。媒介物为酸化水(0.01nhcl,ph~2),并且调整悬浮液的浓度,使得0.35ml悬浮液中包含适当剂量。小鼠的胃容量为大约0.4ml,据认为0.35ml是理想的口服剂量体积,以便不使胃容量超负荷和/或避免食管返流(jpharmpharmacol.2008,60(1),63-70)。悬浮液制备和剂量给药之间的时间间隔为约30秒。给药之后,将小鼠关在笼子中,自由获得水。在给药后2、5、10、15、30、45、60、120和180min从眶窦采集血液样品(~1ml等分试样)。将血液样品离心,并且将血清样品冷藏直至测定。利用immulite
nanoamorphous(a)、microamorphous(b)和结晶状态的卡马西平(c)经180min获得的药代动力学曲线在图11中示出。虚线对应于免疫测定方法的定量极限,其为1.25mg/l。因此,将具有低于所述方法的loq的量的卡马西平的血清样品通过液-液提取法处理并利用hplc测定。将血清的等分试样转移至2ml微量离心管。然后以比例1:4v/v将甲醇添加至每个管并涡旋混合5min。由于水溶性蛋白的沉淀,形成白色不透明溶液。然后将样品以2000rpm离心5min。提取上清液并以低体积插入管(150ul)直接转移至hplc小瓶。利用以前描述的hplc条件分析每个样品。如果认为提取过程的收率为60%,图11中的点划线对应于血清样品中可获得的最大卡马西平浓度。这个平均收率值是通过将提取方法应用于阳性样品(即,高于免疫测定方法的loq的样品)来测定的。
当与microamorphous制剂或结晶药物相比时,用按照本发明的范围制备的nanoamorphous系统观察到小鼠中增强的生物利用度。
观察到的差异可以用制剂之间的粒径差异解释。在暴露于胃肠液时nanoamorphous颗粒的高表面积导致非常快的溶出度,这转而有助于溶液中更大量的卡马西平。
- 还没有人留言评论。精彩留言会获得点赞!