用精氨酸耗竭和免疫肿瘤药剂治疗癌症的组合物和方法与流程
人们认识到某些肿瘤细胞对氨基酸(例如l-精氨酸)有高需求并在l-精氨酸耗竭的条件下被杀死已经有50多年(wheatley和campbell,2002)。在人细胞中,l-精氨酸分三步合成;首先,用鸟氨酸转氨甲酰酶(otc)从l-鸟氨酸和氨基甲酰磷酸合成l-瓜氨酸,精氨基琥珀酸合成酶(ass)将l-瓜氨酸和天冬氨酸转化为精氨基琥珀酸,接着用精氨基琥珀酸裂解酶(asl)将精氨基琥珀酸转化为l-精氨酸和富马酸。大量的肝细胞癌(hcc)、黑素瘤和肾细胞癌(ensoretal.,2002;feunetal.,2007;yoonetal.,2007)不表达ass,因而对l-精氨酸耗竭是敏感的。缺乏ass表达的分子基础似乎是多种多样的,包括异常的基因调控。虽然非恶性细胞在耗尽l-精氨酸时进入静止状态(g0),并因此仍能存活数周,但肿瘤细胞存在细胞周期缺陷,其导致即使蛋白合成受到抑制,也重新启动dna合成,进而导致严重失衡和迅速细胞死亡(shenetal.,2006;scottetal.,2000)。l-精氨酸耗竭对hcc、黑素瘤和其它ass-缺乏癌细胞的选择性毒性已在体外、在异种移植物动物模型中和在临床试验中得到广泛的证明(ensoretal.,2002;feunetal.,2007;shenetal.,2006;izzoetal.,2004)。最近chengetal.(2007)证明许多hcc细胞也缺乏鸟氨酸转氨甲酰酶表达,因此它们也容易受到酶的l-精氨酸耗竭的影响。
人们对使用l-精氨酸水解酶治疗癌症,特别是治疗诸如肝癌、黑素瘤和肾细胞癌的癌症(例如,这些癌症是与高发病率相关的常见癌症形式)感兴趣。两种l-精氨酸降解酶已被用于癌症疗法:细菌精氨酸脱亚胺酶和人精氨酸酶。不幸的是,这两种酶都显示出严重的缺点,其成为临床使用的主要障碍(分别为免疫原性和在血清中具有极差稳定性的低催化活性)。因此,l-精氨酸耗竭疗法的治疗成功将取决于解决这些缺点。
治疗许多癌症的另一个挑战是某些癌症逃避免疫系统的能力。例如,一些肿瘤通过免疫检查点途径来做到这一点,免疫检查点途径是免疫系统中通过调节免疫响应来维持自我耐受性的抑制途径。这些途径可以由导致免疫抵抗的肿瘤失调。这些途径中的一些,无论是促刺激受体的激动剂还是抑制信号的拮抗剂(这两者均导致抗原特异性t-细胞反应的放大),已经成为癌症免疫疗法的靶标。一些示范性受体和配体包括细胞毒性t-淋巴细胞相关抗原4(ctla4)、程序性细胞死亡1(pd1)、程序性细胞死亡配体1(pdl1)、淋巴细胞活化基因3(lag3)、b7-h3、b-7-h4和t细胞膜蛋白3(tim3)等(pardoll,2012)。
发明简述
本公开内容的一个方面一般涉及用耗竭血清中的l-精氨酸的酶来治疗癌症的组合物和方法。在一些实施方案中,癌症是指不表达或以其它方式缺乏精氨基琥珀酸合成酶(ass)、鸟氨酸转氨甲酰酶(otc)或精氨基琥珀酸裂解酶(asl)的癌症。
在一些方面,本发明还涉及其中天然金属辅因子(mn2+)被另一种金属替代的精氨酸酶蛋白的用途。在特定的实施方案中,精氨酸酶蛋白包含人i型精氨酸酶的氨基酸序列或人ii型精氨酸酶的氨基酸序列和非-天然金属辅因子。在一些实施方案中,金属是钴(co2+)。本发明的人i和ii型精氨酸酶蛋白具有两个mn(ii)位点;任一个或两个位点都可以被替代,从而生成具有非-天然金属辅因子的修饰的i或ii型精氨酸酶蛋白。在一些实施方案中,蛋白表现出在ph7.4下大于400mm-1s-1的kcat/km。在一个特定的实施方案中,蛋白表现出在ph7.4下400mm-1s-1和4,000mm-1s-1之间的kcat/km。在另一个实施方案中,蛋白表现出在ph7.4于37℃下400mm-1s-1和2,500mm-1s-1之间的kcat/km。在一个特定的实施方案中,本发明涉及包含人i或ii型精氨酸酶的氨基酸序列和非-天然金属辅因子的蛋白,其中所述蛋白表现出于37℃、ph7.4下大于400mm-1s-1的kcat/km。
本公开内容的又一个方面是通过与例如靶向免疫检查点途径的免疫疗法治疗相结合的精氨酸耗竭来治疗癌症或肿瘤的方法。精氨酸耗竭可以用给予人i或ii型精氨酸酶,包括工程改造或衍生的精氨酸酶,以及来自其它物种的精氨酸酶或其它精氨酸耗竭酶实现,这些酶在与免疫检查点靶向疗法一起给予时表现出至少相加或协同效应。
因此,本公开内容可在某些实施方案中描述为一种在受试者中抑制肿瘤生长的方法,该方法包括给予药物组合物,其包含治疗量的含有钴辅因子的人i型精氨酸酶和免疫-肿瘤药剂。肿瘤可以是多种类型的,它们对精氨酸耗竭疗法有反应,在某些实施方案中,肿瘤是精氨酸营养缺陷型肿瘤,或包括精氨酸依赖型或营养缺陷型细胞。在某些实施方案中,营养缺陷型细胞表现出ass、otc、asl或其组合中一个或多个的表达减少或抑制,因此需要肿瘤细胞利用来自血清的精氨酸。
在某些实施方案中,人i型精氨酸酶或其它酶通过与稳定剂缔合而稳定,以增加在患者血清中酶的半衰期。如本文所用的“缔合”可包括许多缔合类型的任何一种,包括(但不限于)共价或非-共价键,并且也可以包括蛋白融合物,其从工程核酸构建体表达,来自氢键合或疏水性相互作用和本领域技术人员已知的其它方式。用于本公开的方法的稳定剂可包括但不限于聚乙二醇(通常称为聚乙二醇化)、与一种或多种同源合成蛋白聚合物缀合(称为延长化,并可以商品名xten®经市售获得)、与一种或多种fc片段缀合、或与血清蛋白例如白蛋白缀合。所有这样的稳定的酶和本领域技术人员将想到的其它酶均被本公开内容所预期。
本公开的方法可适用于人和非-人动物受试者二者,包括但不限于兽医、农业、家畜或研究动物。本公开内容的一个方面是免疫-肿瘤药剂增强受试者的免疫响应。在某些实施方案中,增强免疫系统包括增加患者对肿瘤存在的t-细胞反应的活性。因此,在某些实施方案中,免疫-肿瘤药剂抑制免疫阻抑物,其有时为细胞表面受体(称为检查点抑制剂),或这样一种受体的配体。实例包括,但不限于pd-1途径抑制剂例如抗-pd-1抗体或抗-pd-l1抗体、ox40(cd134)途径抑制剂例如抗-ox40或抗-ox40l(cd252)、抗-4-1bb或其它抗b7家族配体例如抗-b7-h1和抗-b7.1。示例性抗体包括但不限于派姆单抗(pembrolizumab)、伊匹木单抗、阿特朱单抗(atezolizumab)或纳武单抗(nivolumab)。
本公开内容的方法被构思用于治疗任何有反应的癌症或肿瘤,包括但不限于肝细胞癌、肾细胞癌、乳腺癌、黑素瘤、前列腺癌、胰腺癌、膀胱癌、结肠癌、结肠直肠癌、三阴性乳腺癌、何杰金氏淋巴瘤、胃癌、成胶质细胞瘤、merkel细胞癌、肺癌、小细胞肺癌或非-小细胞肺癌。人i型精氨酸酶和抗-pd-1抗体或抗-pdl-1抗体或其它免疫检查点或tnf受体抑制剂的组合给予与由给予治疗剂量的单独的抗-pd-1抗体或单独的抗-pd-li抗体或单独的人i型精氨酸酶表现出的肿瘤生长抑制比较,可表现出对肿瘤生长抑制的相加效应,或在某些实施方案中,表现出对肿瘤生长或癌症的大于相加的效应,或协同效应。两个治疗方案可以同时施用,或它们可以根据需要依次施用。
本公开内容也可在某些实施方案中描述为一种在癌症患者中治疗癌症的方法,其包括给予所述患者治疗量的包含含有钴辅因子的聚乙二醇化人i型精氨酸酶的药物组合物和包括给予包含免疫-肿瘤药剂的药物组合物的免疫系统调节疗法。
在某些实施方案中,含有钴辅因子的聚乙二醇化人i型精氨酸酶的治疗量是从约0.01mg/kg至约7.5mg/kg,约0.05mg/kg至约5mg/kg,或约0.1mg/kg至约5mg/kg,或从上述范围推导或在上述范围内包含的任何量。
包含含有钴辅因子的聚乙二醇化人i型精氨酸酶的药物组合物可经胃肠外给予,或它可经本领域已知的各种途径递送,包括但不限于局部、皮下、静脉内、皮内、动脉内、腹膜内、病灶内、颅内、关节内、前列腺内、胸膜内、气管内、眼内、鼻内、玻璃体内、阴道内、直肠内、肌内、皮下、结膜下、囊内、粘膜、心包内、脐带内、口服、通过吸入、通过注射、通过输注、通过连续输注、经导管、经灌洗或直接通过局部灌注浸泡靶细胞。在某些实施方案中,药物组合物经静脉内或皮下给予。
本公开内容也可被描述为一种在癌症患者中治疗癌症的方法,其包括给予所述患者精氨酸耗竭剂和检查点途径抑制剂或其它抑制或减少癌症生长或增殖的免疫系统调节剂。该方法还包括治疗癌症,其中与单用精氨酸耗竭剂或单用所述检查点途径抑制剂治疗比较,用精氨酸耗竭剂和检查点途径抑制剂治疗的疗效为相加的,或其中与单用精氨酸耗竭剂或单用所述检查点途径抑制剂治疗比较,用所述精氨酸耗竭剂和检查点途径抑制剂治疗的疗效为协同的。在某些实施方案中,治疗可导致患者中血清精氨酸减少50%-99%或90%-99%,或患者中血清精氨酸减少至不可检测的水平。
可用于实施所述方法的酶可包括精氨酸酶、精氨酸脱亚胺酶或其组合。所述酶可以是人酶、重组人酶、工程改造的人酶或来自其它物种(哺乳动物或者细菌,例如包括但不限于支原体)的酶。
在一些实施方案中,天然精氨酸酶仅通过金属辅因子的替代而被修饰。在其它实施方案中,精氨酸酶除其它修饰,例如替代、缺失、截短或通过与稳定蛋白质或聚合物缀合(例如通过聚乙二醇化)而稳定外,还通过金属辅因子的替代而进行修饰。在一个特定的实施方案中,本发明提供一种包含人i或ii型精氨酸酶的天然氨基酸序列和非-天然金属辅因子的蛋白,其中氨基酸序列缺乏天然序列的一部分。在特定的实施方案中,非-天然金属辅因子是钴。在一些实施方案中,精氨酸酶缺少野生型序列的一部分。在其它实施方案中,氨基酸序列包含截短的i型精氨酸酶或ii型精氨酸酶序列。在一个特定的实施方案中,精氨酸酶是ii型精氨酸酶且缺乏野生型序列的前21个氨基酸。在另一个实施方案中,天然精氨酸酶缺乏n-末端甲硫氨酸。
在另一个方面,本发明涵盖一种包含至少一个氨基酸替代的精氨酸酶蛋白,其中蛋白在生理条件下,特别是在人血清ph(ph7.4)时,与天然人i或ii型精氨酸酶蛋白相比表现出增加的催化活性。在一些实施方案中,精氨酸酶蛋白是人i型精氨酸酶蛋白或人ii型精氨酸酶蛋白。在一些实施方案中,所述蛋白还包含非-天然金属辅因子。在特定的实施方案中,非-天然金属辅因子是co+2。用co+2替代mn+2辅因子导致在生理ph下催化活性的显著增加和km的急剧减少。
在一些方面,本发明还构思了包含连接于非-精氨酸酶氨基酸序列的精氨酸酶的融合蛋白。在一个实施方案中,非-精氨酸酶序列包含免疫球蛋白的fc区域的至少一部分,例如以在给予患者时增加血清中精氨酸酶的半衰期。fc区域或其部分可以是任何合适的fc区域。在一个实施方案中,fc区域或其部分是iggfc区域。在一些实施方案中,具有精氨酸酶活性的氨基酸序列选自人i型精氨酸酶的天然或突变的氨基酸序列和人ii型精氨酸酶的天然或突变的氨基酸序列或本领域已知的其它精氨酸耗竭酶。在某些实施方案中,构思了二聚fc-精氨酸酶融合蛋白、白蛋白或合成蛋白缀合物。
融合蛋白中的精氨酸酶可以是天然的、突变的,和/或其它方式修饰的,例如金属辅因子修饰的。在一些实施方案中,精氨酸酶可含有缺失、替代、截短或其组合。在一个特定的实施方案中,本发明构思了一种含有fc-精氨酸酶的融合蛋白,其中精氨酸酶是i型精氨酸酶。在一个实施方案中,精氨酸酶缺乏一部分野生型序列。在另一个实施方案中,精氨酸酶是缺乏n-末端甲硫氨酸的i型精氨酸酶。在还一个实施方案中,精氨酸酶是ii型精氨酸酶,其中ii型精氨酸酶缺乏野生型ii型精氨酸酶序列的前21个氨基酸。在一些实施方案中,精氨酸酶还包含非-天然金属辅因子。在这些实施方案中,任何一个或两个位点可被替代以生成包含人i或ii型精氨酸酶的氨基酸序列和非-天然金属辅因子的融合蛋白。在一些实施方案中,非-天然金属辅因子是钴。在一些实施方案中,精氨酸酶含有替代。用于本公开内容的示例性精氨酸酶被更全面地描述于美国专利号8,440,184中,其通过引用以其全文结合到本文中。
本发明还构思了通过给予本发明的精氨酸酶蛋白的治疗方法,且特别是治疗癌症受试者的方法。在一些实施方案中,所述癌症是不表达或以其它方式缺乏ass、otc或asl的癌症。在特定的实施方案中,人类癌症是精氨酸营养缺陷型癌症。如上文所讨论的,精氨酸酶蛋白可以是天然的、突变的,和/或以其它方式修饰的,例如金属辅因子修饰的。在一个实施方案中,本发明构思了一种治疗人类癌症患者的方法,其包括给予患者包含融合蛋白的制剂,所述融合蛋白包含具有精氨酸酶活性的氨基酸序列和人免疫球蛋白的fc区域的至少一部分。在一些实施方案中,在癌症的至少一部分癌细胞被杀死的情况下进行给药。在另一个实施方案中,所述制剂包含氨基酸序列,所述氨基酸序列具有比真人精氨酸酶在生理条件下所显示的更高的人精氨酸酶活性,并且还包含一个或多个附加的聚乙二醇链。在一些实施方案中,制剂是包含任何上文讨论的精氨酸酶蛋白和药学上可接受的赋形剂的药物制剂。这样的药学上可接受的赋形剂是本领域技术人员熟知的。全部以上精氨酸酶变体均被构思为可用于人类疗法。
癌症可以是任何类型的癌症或肿瘤类型。在一些实施方案中,癌症是肝细胞癌、肾细胞癌、黑素瘤、前列腺癌或胰腺癌。在一些实施方案中,制剂经局部、静脉内、皮内、动脉内、腹膜内、病灶内、颅内、关节内、前列腺内、胸膜内、气管内、眼内、鼻内、玻璃体内、阴道内、直肠内、肌内、皮下、结膜下、囊内、粘膜、心包内、脐带内、口服、通过吸入、通过注射、通过输注、通过连续输注、经导管、经灌洗或直接通过局部灌注浸泡靶细胞来给予。
在一个优选的实施方案中,所有上述的精氨酸酶、变体等被预期为纯化或分离的蛋白,并且优选是单体蛋白。
在实施例章节中的实施方案被理解为是可适用于本发明所有方面的本发明的实施方案。
权利要求书中使用术语“或”被用来意指“和/或”,除非明确表示仅指可选方案,或可选方案相互排斥,尽管本公开内容支持仅提及可选方案和“和/或”的定义。
在整个本申请中,术语“约”被用来表示一个值包括用于确定该值的组合物、装置或方法的误差的标准偏差。
根据存在已久的专利法,在权利要求书或说明书中,单词“一”和“一个”在与单词“包含”结合使用时表示一个或多个,除非特别注明。
如本文所用的术语“治疗有效的”指本发明的方法中使用以达到治疗效果的活性剂和/或治疗组合物(例如治疗性多核苷酸和/或治疗性多肽)的量,例如,其中正在治疗的病症的至少一个症状至少被改善,和/或与这些细胞结合使用的过程或材料的分析。
本发明的其它目的、特征和优点将从以下详细描述中而变得显而易见。然而,应该理解,详细的描述和具体的实例,虽然指出了本发明的具体实施方案,但只是为了举例说明,因为对本领域技术人员而言,在本发明的精神和范围内的各种变化和修改将从该详细的描述而变得显而易见。
附图简述
下列附图构成本说明书的一部分,并包括这些附图,以进一步显示本发明的某些方面。通过参考这些附图中的一个或多个,结合本文所呈现的具体实施方案的详细描述,可以更好地理解本发明。
图1是显示在小鼠模型中血清l-精氨酸耗竭的图。用单个ip剂量的co-hargi治疗的balb/c小鼠中血清l-arg浓度保持≤3-4µm超过3天。
图2是显示与对照相比,在用co-hargi治疗时hcc肿瘤异种移植物减少的图。荷载hep3b肿瘤异种移植物的裸小鼠在第9天和第12天经ip注射pbs(◦)或co-hargi(●)治疗两次。用co-hargi治疗的小鼠中观察到肿瘤缩小,而pbs治疗的肿瘤生长未受抑制。
图3是显示钴负载对人i型精氨酸酶的催化活性的影响的图。
图4是显示在ct26小鼠模型中,用co-hargi、抗pd-l1和co-hargi和抗pd-l1的组合抑制结肠癌肿瘤生长的图。如数据所示,2种药剂的组合对抑制肿瘤生长具有大于相加效应。
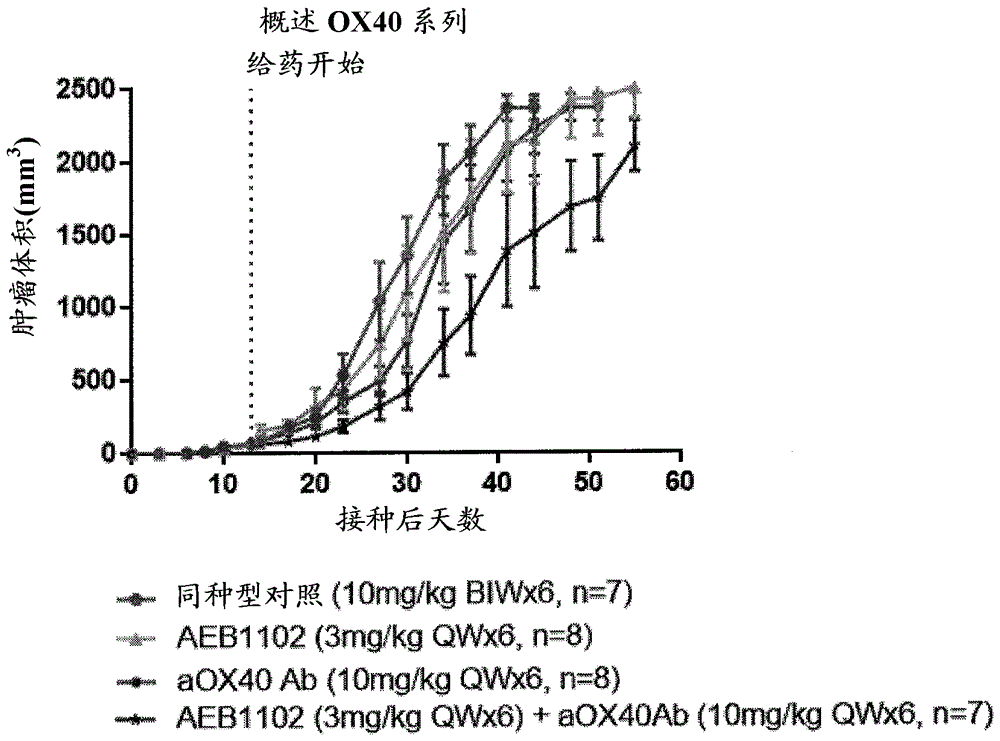
图5是显示在mc38小鼠模型中,用co-hargi、抗ox40抗体,和co-hargi和抗ox40抗体的组合抑制结肠癌肿瘤生长的图。如数据所示,2种药剂的组合对抑制肿瘤生长具有大于相加效应。
图6是显示用co-hargi、抗pd-l1以及co-hargi和抗pd-l1的组合,ct26小鼠模型中存在的cd45+t细胞的图。
图7是显示如图6所示的cd45+t细胞中存在的cd8+细胞%的图。
示例性实施方案的描述
本发明一般涉及用耗竭血清中l-精氨酸的酶治疗癌症的组合物和方法。在一些实施方案中,癌症是不表达或以其它方式缺乏精氨基琥珀酸合成酶(ass)、鸟氨酸转氨甲酰酶(otc)或精氨基琥珀酸裂解酶(asl)或精氨酸生物合成所需的其它酶的癌症。考虑了天然酶和突变酶二者,以及含有修饰的金属辅因子的酶,与其它多肽融合的酶,以及与提高血清持久性的聚合物(例如高分子量聚乙二醇)缀合的酶。
i.精氨酸酶
精氨酸酶是含有锰的酶。它是尿素循环的最终酶。精氨酸酶是尿素循环的第五步,也是最后一步,尿素循环是在哺乳动物中身体处置有害氨的一系列生物物理反应。特别地,精氨酸酶将l-精氨酸转变成l-鸟氨酸和尿素。
l-精氨酸是一氧化氮合酶(nos)的供氮底物,产生l-瓜氨酸和no。虽然有报道称,对l-精氨酸,精氨酸酶的km(2-5mm)比nos(2-20µm)高得多,但精氨酸酶也可能在调节nos活性方面起作用。在某些条件下,i型精氨酸酶被cys-s-亚硝基化,导致对l-精氨酸的亲和力更高和nos的底物可用性减少。
精氨酸酶是一种具有被几个螺旋围绕的平行八股β片的α/β折叠的均三聚酶。该酶含有一个双核金属簇,它是生成氢氧化物以对l-精氨酸的胍鎓碳进行亲核攻击所整体必需的。用于精氨酸酶的天然金属是mn2+。这些mn2+离子配位水,使分子取向和稳定,并使水用作亲核试剂并攻击l-精氨酸,使其水解为鸟氨酸和尿素。
哺乳动物有两种精氨酸酶同工酶(ec3.5.3.1),其催化l-精氨酸水解为尿素和l-鸟氨酸。i型精氨酸酶基因位于染色体6(6q.23),在肝细胞胞质中高度表达,并作为尿素循环的最后一步起着除去氮的作用。ii型精氨酸酶基因被发现在染色体14(14q.24.1)上。ii型精氨酸酶以线粒体位于组织例如肾、脑和骨骼肌中,认为在这些组织中它为脯氨酸和聚胺生物合成提供l-鸟氨酸供给(lopezetal.,2005)。
精氨酸酶作为一种降解细胞外l-精氨酸的方法已研究了近50年(dillonetal.,2002)。通过经肝动脉栓塞技术引入精氨酸酶,已取得一些有前景的临床结果;此后,几名患者经历了hcc的部分缓解(chengetal.,2005)。然而,因为精氨酸酶具有高的km(~2-5mm)并在生理ph值下表现出很低的活性,为了化疗目的需要高剂量(dillonetal.,2002)。虽然在几分钟内从循环清除天然精氨酸酶(savocaetal.,1984),但在大鼠内单次注射peg-精氨酸酶mw5000已足以实现精氨酸的几乎完全耗尽,持续约3天(chengetal.,2007)。
chengetal.令人惊讶地观察到,许多人hcc细胞系不表达otc(除了ass外),因此它们容易受到peg-精氨酸酶的影响(chengetal.,2007)。在植入hep3b肝癌细胞的小鼠中,每周给予peg-精氨酸酶导致肿瘤生长迟缓,其通过联合给予5-氟尿嘧啶(5-fu)而增强。然而,peg-精氨酸酶以不适用于人治疗的极高剂量使用,反映了其较低的生理活性。
为解决这些问题,显示良好的动力学和稳定性的细菌精氨酸水解酶,精氨酸脱亚胺酶或adi已在体外和临床上进行了测试。不幸的是,adi是一种细菌酶,因此它在大多数患者中引起强烈的免疫反应和不良反应。然而,对于那些没有出现重大不良反应的患者,相当大比例的患者表现出稳定的疾病或缓解。
对于临床应用来说,精氨酸酶经工程改造以允许它在循环中持续很长的时间(例如,几天)是必要的。在没有任何修饰的情况下,人精氨酸酶在循环中仅具有几分钟的半衰期,这主要是因为它的大小不足以大到避免过滤通过肾脏。未修饰的人精氨酸酶在血清中极易被失活,其降解的半衰期仅为4小时。因此,本发明为临床研究和潜在治疗用途开发了具有改善循环持久性的新且改进形式的精氨酸酶。
ii.精氨酸酶变体
哺乳动物具有两种催化l-精氨酸水解为尿素和l-鸟氨酸的精氨酸酶同工酶(ec3.5.3.1)。i型精氨酸酶基因位于染色体6(6q.23)上,在肝细胞胞质中高度表达,并作为尿素循环的最后一步起着除去氮的作用。ii型精氨酸酶基因被发现在染色体14(14q.24.1)上,ii型精氨酸酶以线粒体位于组织例如肾、脑和骨骼肌中,在这些组织中它被认为为脯氨酸和聚胺生物合成提供了l-鸟氨酸供给(lopezetal.,2005)。
l-精氨酸是是一氧化氮合酶(nos)的唯一底物,产生l-瓜氨酸和no。虽然有报道称,对于l-精氨酸,精氨酸酶的km(2-5mm)比nos(2-20µm)高得多,但精氨酸酶也可能在调节nos活性方面起作用(duranteetal.,2007)。在某些条件下,i型精氨酸酶被cys-s-亚硝基化,导致对l-精氨酸的亲和力提高和nos的底物的可用性减少(santhanametal.,2007)。精氨酸酶是一种具有被几个螺旋围绕的平行八股β片的α/β折叠的均三聚酶。该酶含有一个双核金属簇,它是生成氢氧化物以对l-精氨酸的胍鎓碳进行亲核攻击所整体必需的(camaetal.,2003;dowlingetal.,2008)。用于精氨酸酶的天然金属是mn2+。具有天然金属(即mn2+)的精氨酸酶表现出9的ph最佳值。在生理ph下,所述酶在l-精氨酸水解过程中表现出低于1/10的kcat/km。由具有天然mn2+酶的真人精氨酸酶显示的低催化活性给人类治疗带来了问题,因为这意味着可能必须使用该酶的不切实际的剂量以达到l-精氨酸血浆水平的治疗性相关降低。
在一些方面,本发明构思了突变体精氨酸酶,其中天然金属辅因子(mn2+)被另一种金属替代。已发现当与具有天然金属辅因子的天然人精氨酸酶比较时,人精氨酸酶中的金属辅因子的替代对在生理条件下l-精氨酸水解速率和稳定性发挥有益的作用。用其它二价阳离子替代天然金属(mn2+)可被用来将酶的最适ph值转换为较低的值,从而在生理条件下达到高的l-精氨酸水解速率。本发明的人i和ii型精氨酸酶蛋白具有两个mn(ii)位点;因此,任何一个或两个位点可被替代以产生具有非天然金属辅因子的突变i或ii型精氨酸酶蛋白。
在一些实施方案中,金属是钴(co2+)。掺入co2+以替代人i型精氨酸酶或人ii型精氨酸酶中的mn2+导致在生理ph下显著更高的活性。已发现与含有mn2+的人i型精氨酸酶(“mn-hargi”)比较,含有co2+的人i型精氨酸酶(“co-hargi”)显示出在ph7.4时体外kcat/km的10倍增加,这进而转化为hcc细胞毒性的15倍增加和黑素瘤细胞毒性的13倍增加。还发现单次注射co-hargi的药物制剂可清除小鼠血清l-arg超过3天。此外,已发现co-hargi的药物制剂可以使裸鼠内的hcc肿瘤异种移植物缩小,而mn-hargi只延缓肿瘤的生长(ensoretal.,2002)。
在本发明的某些方面,公开了涉及聚乙二醇化精氨酸酶的方法和组合物。特别地,在工程化半胱氨酸残基(例如,替代n-末端的第三残基)处使精氨酸酶聚乙二醇化,可用来生产同质聚乙二醇化精氨酸酶组合物。用于基于聚合反应暂时中断分离聚乙二醇化精氨酸酶的方法也被公开。
聚乙二醇化是聚(乙二醇)聚合物链共价连接于另一个分子(通常为药物或治疗性蛋白)的过程。通过将peg的反应性衍生物与目标大分子一起孵育,常规地实现了聚乙二醇化。peg共价连接于药物或治疗性蛋白可“掩盖”所述药剂免于宿主的免疫系统(减少免疫原性和抗原性),增加药剂的流体力学尺寸(溶液中的尺寸),这通过降低肾清除率来延长其循环时间。聚乙二醇化也可为疏水性药物和蛋白质提供水溶性。
聚乙二醇化中的第一步是peg聚合物在一个或两个末端的合适的官能化。在每个末端用相同的反应性部分激活的peg被称为“同双官能的”,而如果存在的官能团是不同的,则将peg衍生物称为“异双官能的”或“异官能的”。制备peg聚合物的化学活性或活化的衍生物以将peg连接至所需分子。
用于peg衍生物的合适官能团的选择根据将偶联到peg的分子上可利用的反应基团的类型进行。对于蛋白,典型的反应性氨基酸包括赖氨酸、半胱氨酸、组氨酸、精氨酸、天冬氨酸、谷氨酸、丝氨酸、苏氨酸、酪氨酸。也可使用n-末端氨基和c-末端羧酸。
用于形成第一代peg衍生物的技术通常是使peg聚合物和与羟基具有反应性的基团反应,所述基团通常是酸酐、酰氯、氯甲酸酯和碳酸酯。在第二代聚乙二醇化化学中,更有效的官能团如醛、酯、酰胺等可用于缀合。
随着聚乙二醇化的应用越来越先进和复杂,对于用于缀合的异双官能的peg的需求日益增加。这些异双官能的peg在连接两个实体中是非常有用的,其中需要亲水、柔韧和生物相容的间隔基。对于异双官能的peg的优选端基是马来酰亚胺、乙烯砜、吡啶基二硫化物、胺、羧酸和nhs酯。
最常见的修饰剂,或接头,以甲氧基peg(mpeg)分子为基础。它们的活性取决于在醇端加入蛋白-修饰基团。在某些情况下,聚乙二醇(peg二醇)被用作前体分子。随后二醇在两端被修饰,以制备异或同二聚peg-连接的分子(如在实施例中用peg双-乙烯砜显示)。
蛋白质一般都是在亲核位点例如未质子化硫醇(半胱氨酰残基)或氨基进行聚乙二醇化。半胱氨酰-特异性修饰试剂的实例包括peg马来酰亚胺、peg碘醋酸酯、peg硫醇和peg乙烯砜。在温和的条件和中性到略碱性的ph下,所有这4种都是半胱氨酰强特异性的,但各有一些缺点。在碱性条件下,由马来酰亚胺形成的酰胺可能有些不稳定,因此在使用该接头的制剂选择上可能存在一定的局限性。用碘代peg形成的酰胺键是更稳定的,但游离碘可在一些条件下修饰酪氨酸残基。peg硫醇与蛋白硫醇形成二硫键,但在碱性条件下,这种键也可能是不稳定的。peg-乙烯砜与马来酰亚胺和碘代peg相比,反应性是相对慢的;然而,形成的硫醚键是相当稳定的。其较慢的反应速度也能使peg-乙烯砜反应更容易控制。
在天然半胱氨酰残基上的位点-特异性聚乙二醇化很少进行,因为这些残基通常是以二硫键的形式存在,或者是生物活性所必需的。另一方面,定点诱变可用于掺入对于硫醇特异性接头的半胱氨酰聚乙二醇化位点。半胱氨酸突变必须被设计为使得其可接近聚乙二醇化试剂并且在聚乙二醇化之后仍然是生物活性的。
胺-特异性修饰剂包括pegnhs酯、peg三氟乙磺酸酯、peg醛、peg异硫氰酸酯和几种其它修饰剂。它们都在温和的条件下反应,对氨基是非常特异性的。pegnhs酯可能是较具反应性的试剂之一,然而,其高反应性可能使聚乙二醇化反应难以大规模控制。peg醛与氨基形成亚胺,其然后用氰基硼氢化钠还原成仲胺。与硼氢化钠不同,氰基硼氢化钠不会还原二硫键。然而,这种化学物质毒性很高,必须小心地加以处理,特别是在它易挥发的较低ph下。
由于在大多数蛋白质上的多个赖氨酸残基,位点-特异性聚乙二醇化可能是一个挑战。幸运的是,由于这些试剂与未质子化的氨基发生反应,所以通过在较低的ph下进行反应,有可能将聚乙二醇化导向较低pk的氨基。一般来说,α-氨基的pk比赖氨酸残基的ε-氨基的pk低1-2个ph单位。通过使分子在ph7或以下聚乙二醇化,可频繁地实现对n-末端的高选择性。然而,只有当对于生物活性不需要蛋白质的n-末端部分时,这才是可行的。尽管如此,来自聚乙二醇化的药代动力学获益经常超过体外生物活性的显著损失,导致具有更大的体内生物活性的产物,而与聚乙二醇化化学无关。
当开发聚乙二醇化过程时需要考虑几个参数。幸运的是,通常不超过4个或5个关键参数。对聚乙二醇化条件进行优化的“实验设计”方法可能是非常有用的。对于硫醇-特异性聚乙二醇化反应,要考虑的参数包括:蛋白浓度、peg与蛋白比率(基于摩尔计)、温度、ph、反应时间,和在某些情况下,排除氧。氧可以促进蛋白的分子间二硫化物生成,从而降低聚乙二醇化产物的产率。对胺-特异性修饰应考虑相同的因素(除氧外),除了ph可能是甚至更关键的,特别是在针对n-末端氨基时。
对于胺-和硫醇-特异性修饰,反应条件可影响蛋白的稳定性。这可限制温度、蛋白浓度和ph。此外,在开始聚乙二醇化反应之前应了解peg接头的反应性。例如,如果聚乙二醇化试剂仅为70%活性的,所用peg的量应确保只有活性peg分子在蛋白与peg反应化学计量中被计算。如何确定peg反应性和质量将在后面描述。
iv.蛋白和肽
在某些实施方案中,本发明涉及包含至少一种蛋白或肽(例如稳定的精氨酸酶多聚体)的新的组合物。这些肽可包含在融合蛋白中或与如上所述的试剂缀合。
a.蛋白和肽
如本文所用的,蛋白或肽通常指,但不限于,大于约200个氨基酸,直至从基因翻译的全长序列的蛋白;大于约100个氨基酸的多肽;和/或从约3至约100个氨基酸的肽。为了方便起见,术语“蛋白”、“多肽”和“肽”在此可互换使用。
在某些实施方案中,至少一种蛋白或肽的大小可包含,但不限于,1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、约110、约120、约130、约140、约150、约160、约170、约180、约190、约200、约210、约220、约230、约240、约250、约275、约300、约325、约350、约375、约400、约425、约450、约475、约500、约525、约550、约575、约600、约625、约650、约675、约700、约725、约750、约775、约800、约825、约850、约875、约900、约925、约950、约975、约1000、约1100、约1200、约1300、约1400、约1500、约1750、约2000、约2250、约2500个或更多个氨基酸残基。
如本文所用的,“氨基酸残基”指本领域已知的任何天然存在的氨基酸、任何氨基酸衍生物或任何氨基酸模拟物。在某些实施方案中,蛋白或肽的残基是连续的,没有中断氨基酸残基序列的任何非-氨基酸。在其它实施方案中,序列可包含一个或多个非-氨基酸部分。在特定的实施方案中,蛋白或肽的残基的序列可被一个或多个非-氨基酸部分中断。
因此,术语“蛋白或肽”涵盖这样的氨基酸序列,其包含天然存在的蛋白中发现的20个常见氨基酸的至少一个,或至少一个修饰的或不常见的氨基酸,包括但不限于下表1中所示的那些氨基酸。
表1
蛋白或肽可通过本领域技术人员已知的任何技术制得,包括通过标准分子生物技术表达蛋白、多肽或肽,从天然来源分离蛋白或肽,或化学合成蛋白或肽。与各种基因相对应的核苷酸和蛋白、多肽和肽序列之前已被公开,并可在本领域普通技术人员已知的计算机数据库中找到。一个这样的数据库是nationalcenterforbiotechnologyinformation’sgenbankandgenpept数据库(可在网站ncbi.nlm.nih.gov/上获得)。已知基因的编码区域可以使用本文公开的或者对本领域普通技术人员来说是已知的技术进行扩增和/或表达。或者,蛋白质、多肽和肽的各种商业制剂是本领域技术人员已知的。
b.核酸和载体
在本发明的某些方面,编码作为稳定的多聚精氨酸酶的融合蛋白的核酸序列可被公开。取决于要使用的表达系统,可根据常规方法来选择核酸序列。例如,人i和ii型精氨酸酶包含在大肠杆菌中很少使用的可干扰表达的多个密码子,因此其各自的基因或变体可以经密码子优化用于大肠杆菌表达。各种载体也可用来表达感兴趣的蛋白,例如融合多聚精氨酸酶或半胱氨酸-替代的精氨酸酶。示例性载体包括,但不限于,质粒载体、病毒载体、转座子或基于脂质体的载体。
c.宿主细胞
可用于本发明的宿主细胞,优选真核细胞,是可经转化以允许表达和分泌精氨酸酶及其融合多聚体的任何细胞。宿主细胞可以是细菌、哺乳动物细胞、酵母或丝状真菌。各种细菌包括埃希氏菌属(escherichia)和芽胞杆菌属(bacillus)。属于酵母属(saccharomyces)、克鲁维酵母属(kluyveromyces)、汉逊酵母属(hansenula)或毕赤酵母属(pichia)的酵母发现可用作合适的宿主细胞。丝状真菌的各个物种可用作表达宿主,包括以下属:曲霉属(aspergillus)、木霉属(trichoderma)、链孢霉属(neurospora)、青霉属(penicillium)、头孢霉属(cephalosporium)、绵霉属(achlya)、足孢子菌属(podospora)、内座壳属(endothia)、毛霉属(mucor)、旋孢腔菌属(cochliobolus)和梨孢霉属(pyricularia)。
可用的宿主生物的实例包括细菌,例如大肠杆菌mc1061、枯草芽胞杆菌brb1的衍生菌(sibakovetal.,1984)、金黄色葡萄球菌sai123(lordanescu,1975)或streptococcuslividans(hopwoodetal.,1985);酵母,例如酿酒酵母ah22(melloretal.,1983)和粟酒裂殖酵母(schizosaccharomycespombe);丝状真菌,例如构巢曲霉(aspergillusnidulans)、泡盛曲霉(aspergillusawamori)(ward,1989)、里氏木霉(penttilaetal.,1987;harkkietal,1989)。
哺乳动物宿主细胞的实例包括中国仓鼠卵巢细胞(cho-k1;atccccl61)、大鼠垂体细胞(gh1;atccccl82)、helas3细胞(atccccl2.2)、大鼠肝细胞瘤细胞(h-4-ii-e;atcccrl1548)、sv40-转化猴肾细胞(cos-1;atcccrl1650)和鼠胚胎细胞(nih-3t3;atcccrl1658)。前文是说明性的,但不限于本领域已知的许多可能的宿主生物。原则上,所有能够分泌的宿主都可以使用,无论是原核生物还是真核生物。
表达精氨酸酶和/或它们的融合多聚体的哺乳动物宿主细胞在通常用来培养亲本细胞系的条件下培养。一般来说,细胞在含有生理盐和营养素的标准培养基中培养,例如标准rpmi、mem、imem或dmem,通常补充有5-10%血清(如胎牛血清)。培养条件也是标准的,例如培养物于37℃在固定或滚动培养中孵育,直到达到所需的蛋白水平。
d.蛋白纯化
蛋白纯化技术是本领域技术人员熟知的。这些技术在一个水平上涉及细胞、组织或器官的均质化和粗分级分离为多肽和非-多肽部分。感兴趣的蛋白或多肽可使用色谱和电泳技术进一步纯化,以达到部分或完全纯化(或纯化至均质性),除非另有规定。特别适合于制备纯肽的分析方法有离子交换色谱法、凝胶排阻色谱法、聚丙烯酰胺凝胶电泳法、亲和色谱法、免疫亲和色谱法和等电聚焦法。纯化肽的一种特别有效的方法是快速液相色谱法(fplc),或甚至是高效液相色谱法(hplc)。
纯化的蛋白质或肽意图指可从其它组分中分离出来的组合物,其中蛋白质或肽被纯化至与其自然可获得的状态有关的任何程度。因此,分离的或纯化的蛋白质或肽也指游离于其可自然存在的环境的蛋白质或肽。一般来说,“纯化的”是指一种蛋白质或肽组合物,该蛋白质或肽组合物经过分级分离以去除各种其它组分,并且该组合物基本上保留了其表达的生物活性。在使用术语“基本纯化”时,这个称谓是指这样的组合物,其中蛋白质或肽形成该组合物的主要组分,例如构成该组合物中约50%、约60%、约70%、约80%、约90%、约95%或更多的蛋白质。
适合用于蛋白纯化的各种技术是本领域技术人员熟知的。这些包括,例如,用硫酸铵、peg、抗体等沉淀,或通过加热变性,然后是:离心;色谱步骤例如离子交换、凝胶过滤、反相、羟磷灰石和亲和色谱;等电聚焦;凝胶电泳;以及这些技术和其它技术的组合。如本领域一般已知的,认为进行各种纯化步骤的顺序可以改变,或者可以省略某些步骤,并且仍然导致制备基本纯化的蛋白质或肽的合适方法。
根据本公开的内容,本领域技术人员已知用于量化蛋白质或肽纯化程度的各种方法。这些包括例如确定活性流分的比活性,或通过sds/page分析评估流分内多肽的量。评估流分纯度的一种优选的方法是计算该流分的比活性,将其与初始提取物的比活性进行比较,从而计算其中的纯度,其用“-倍数纯化数”来评估。当然,用来代表活性量的实际单位将取决于在纯化后选择的特定检测技术,以及所表达的蛋白质或肽是否表现出可检测的活性。
没有蛋白或肽总是以其最纯净的状态提供的一般要求。实际上,可以预期,在某些实施方案中,较低基本纯化的产物可具有效用。部分纯化可以通过组合使用较少的纯化步骤或通过使用相同的一般纯化方案的不同形式来实现。例如,应当理解,利用hplc设备进行的阳离子交换柱色谱通常将导致比使用低压色谱系统的相同技术更高“-倍数”的纯化。表现出相对纯化程度较低的方法可在蛋白质产物的总回收率或维持所表达蛋白的活性方面具有优势。
在某些实施方案中,蛋白或肽可以被分离或纯化为例如,稳定的精氨酸酶多聚融合蛋白,或者前或后聚乙二醇化的精氨酸酶。例如,his标签或亲和表位可以包含在这样的精氨酸酶变体中,以便于纯化。亲和色谱是依赖于要分离的物质和它能与之特异性结合的分子之间的特定亲和力的色谱过程。这是一种受体-配体类型的相互作用。所述柱材料通过将结合配偶体之一共价偶联到不溶性基质来合成。然后,柱材料能够从溶液中特定地吸附所述物质。洗脱通过改变条件为不发生结合的条件(例如,改变ph、离子强度、温度等)而发生。基质应该是本身不吸附分子至任何显著程度,并且具有广泛范围的化学、物理和热稳定性的物质。配体应以不影响其结合性质的方式偶联。配体也应该提供相对紧密的结合。而且应该可以在不破坏样品或配体的情况下洗脱该物质。
尺寸排阻色谱法(sec)是一种色谱方法,其中溶液中的分子根据它们的大小,或者用更专业的术语来说,它们的流体力学体积来分离。它通常应用于大的分子或大分子复合物,例如蛋白和工业聚合物。典型地,当用水溶液将样品输送通过柱时,这种技术称为凝胶过滤色谱法,而将有机溶剂用作流动相时,则称为凝胶渗透色谱法。
sec的基本原理是不同尺寸的粒子以不同的速率通过固定相洗脱(过滤)。这导致了基于尺寸的粒子溶液的分离。如果所有的粒子同时或接近同时加载,则相同尺寸的粒子应该一起洗脱。每个尺寸排阻柱都有一个可以分离的分子量范围。排阻极限定义了这个范围的上端的分子量,并且是分子太大而不能在固定相中捕获的情况。渗透极限定义了分离范围的下端的分子量,并且是尺寸足够小的分子能够完全渗透到固定相的孔中的情况,并且这个分子质量以下的所有分子太小,以至于它们作为单一条带洗脱。
高效液相色谱(或高压液相色谱,hplc)是一种在生物化学和分析化学中经常用于分离、鉴定和量化化合物的柱色谱形式。hplc使用容纳色谱填料(固定相)的柱、使流动相通过该柱的泵和显示分子的保留时间的检测器。保留时间取决于固定相、被分析的分子和使用的溶剂之间的相互作用而变化。
v.药物组合物
设想本发明的新精氨酸酶可在具有局部晚期或转移性癌症的癌症患者中全身或局部给予,以抑制肿瘤细胞生长,且最优选地,杀死癌细胞。它们可以经静脉内、鞘内和/或腹膜内给药。它们可单独或与抗-增殖药物组合给予。在一个实施方案中,给予它们以在手术或其它程序前减轻患者中的癌症负荷。或者,它们可以在手术后给予,以确保任何残留的癌症(如手术未能消除的癌症)不能存活。
本发明无意受到治疗制剂的特殊性质的限制。例如,这样的组合物可在制剂中与生理学上可耐受的液体、凝胶或固体载体、稀释剂和赋形剂一起提供。这些治疗制剂可给予哺乳动物以供兽医应用,如家畜,和以类似于其它治疗剂的方式临床应用于人类。一般来说,治疗效果所需的剂量将根据使用的类型和给药方式,以及个体受试者的特殊要求而有所不同。
这样的组合物通常作为可注射的液体溶液或混悬液制备。合适的稀释剂和赋形剂是,例如,水、盐水、葡萄糖、甘油等,及其组合。此外,如果需要,组合物可含有微量辅助物质,例如湿润剂或乳化剂、稳定剂或ph缓冲剂。
在考虑临床应用的情况下,可能需要以适合预期应用的形式制备药物组合物——表达载体、病毒储库、蛋白、抗体和药物。一般来说,本发明的药物组合物包含溶解或分散在药学上可接受的载体中的有效量的一种或多种精氨酸酶变体或其它药剂。短语“药学上或药理学上可接受的”指的是当给予动物(例如人,视情况而定)时不产生不利、过敏或其它不良反应的分子实体和组合物。含有至少一种精氨酸酶变体,例如用本文公开的方法分离的稳定的多聚精氨酸酶或聚乙二醇化精氨酸酶,或其它活性成分的药物组合物的制备将是本领域技术人员根据本公开内容已知的,如通过remington’spharmaceuticalsciences,第18版,1990中举例说明的,其通过引用并入本文。此外,对于动物(如,人)给药,应理解的是,制剂应符合fda生物标准办公室要求的无菌、致热原性、一般安全和纯度标准。
如本文所用的,“药学上可接受的载体”包括任何和所有溶剂、分散介质、包衣料、表面活性剂、抗氧化剂、防腐剂(如,抗细菌剂、抗真菌剂)、等渗剂、吸收延迟剂、盐、防腐剂、药物、药物稳定剂、凝胶、粘合剂、赋形剂、崩解剂、润滑剂、甜味剂、矫味剂、染料等材料及其组合,如为本领域普通技术人员已知的(参见例如,remington’spharmaceuticalsciences,第18版,1990,通过引用并入本文)。除非任何常规载体与活性成分不相容,否则考虑其在药物组合物中的应用。
本发明可包含不同类型的载体,取决于它以固体、液体还是气雾剂的形式给予,以及在这样的注射给药途径中是否需要无菌。本发明可经静脉内、皮内、经皮、鞘内、动脉内、腹膜内、鼻内、阴道内、直肠内、局部、肌内、皮下、粘膜、口服、局部(topically)、局部地(locally)、吸入(如,气雾剂吸入)、注射、输注、连续输注、经导管、经灌洗、直接局部灌注浸泡靶细胞、在脂质组合物(如,脂质体)中,或通过其它方法或前述方法的任何组合给予,如本领域普通技术人员已知的(见,例如,remington’spharmaceuticalsciences,第18版,1990,通过引用并入本文)。
精氨酸酶变体可配制成游离碱、中性或盐的形式的组合物。药学上可接受的盐,包括酸加成盐,例如与蛋白质成分的游离氨基形成的那些,或与无机酸例如盐酸或磷酸,或有机酸例如乙酸、草酸、酒石酸或扁桃酸形成的酸加成盐。与游离羧基形成的盐也可从无机碱例如,钠、钾、铵、钙或铁的氢氧化物;或有机碱例如异丙基胺、三甲胺、组氨酸或普鲁卡因衍生而来。在配制时,溶液将以与剂量配制相容的方式给予,并以治疗有效的量给予。所述制剂容易地以多种剂型给药,例如,为胃肠外给予配制的剂型,例如注射溶液,或递送至肺的气雾剂,或为消化道给予配制的剂型,例如药物释放胶囊等。
此外,根据本发明,适合于给予的本发明的组合物在药学上可接受的载体(有或没有惰性稀释剂)中提供。载体应是可同化的并包括液体、半固态即糊状物,或固体载体。除非任何常规介质、试剂、稀释剂或载体对接受者或其中所含组合物的治疗效果有害,否则其在可给予的组合物中用于实践本发明的方法是适当的。载体或稀释剂的实例包括脂肪、油、水、盐水溶液、脂质、脂质体、树脂、粘合剂、填充剂等,或其组合。组合物也可包含各种减缓一种或多种组分的氧化的抗氧化剂。此外,预防微生物的作用可通过防腐剂产生,例如各种抗细菌剂和抗真菌剂,包括但不限于对羟苯甲酸酯类(如,对羟苯甲酸甲酯、对羟苯甲酸丙酯)、氯丁醇、苯酚、山梨酸、硫柳汞或其组合。
根据本发明,组合物与载体以任何方便和实用的方式,即通过溶解、悬浮、乳化、混合、封装、吸收等结合。这样的程序对本领域技术人员而言是常规的。
在本发明的特定的实施方案中,组合物与半-固体或固体载体结合或充分混合。混合可以任何方便的方式进行,例如研磨。混合过程中也可以添加稳定剂,以防止组合物失去治疗活性,即在胃中的变性。用于组合物的稳定剂的实例包括缓冲剂、氨基酸例如甘氨酸和赖氨酸、碳水化合物例如葡萄糖、甘露糖、半乳糖、果糖、乳糖、蔗糖、麦芽糖、山梨醇、甘露醇等。
在进一步的实施方案中,本发明可涉及药物脂质媒介组合物的使用,这些组合物包括精氨酸酶变体、一种或多种脂质和水性溶剂。如本文所用的,术语“脂质”将被定义为包括任何广泛范围的物质中的任何一种,其在特征上不溶于水并可用有机溶剂提取。这样的广泛种类的化合物是本领域技术人员熟知的,当术语“脂质”在此使用时,它不限于任何具体的结构。实例包括含有长链脂族烃及其衍生物的化合物。脂质可以是天然存在的或合成的(即,由人设计或生产)。然而,脂质通常是一种生物物质。生物脂质是本领域熟知的,并包括例如,中性脂肪、磷脂、磷酸甘油酯、类固醇、萜烯、血溶性脂类、鞘糖脂、糖脂、硫脂、含醚和酯键的脂肪酸的脂类和可聚合脂质,及其组合。当然,本领域技术人员理解为脂质的并非在此具体描述的化合物也被本发明的组合物和方法所涵盖。
本领域普通技术人员将熟悉可用于使组合物分散于脂质媒介中的各种技术。例如,稳定的多聚或聚乙二醇化精氨酸酶可通过本领域普通技术人员所知的任何手段,分散在含有脂质的溶液中,用脂质溶解,用脂质乳化,与脂质混合,与脂质结合,共价结合于脂质,作为悬浮液包含在脂质中,包含在胶束或脂质体中或与胶束或脂质体复合,或以其它方式与脂质或脂质结构缔合。分散可能导致或可能不导致脂质体的形成。
给予动物患者的本发明组合物的实际剂量,可根据身体和生理因素来确定,例如体重、病情的严重程度、正在治疗的疾病类型、先前或同时进行的治疗干预、患者的原发症和给药途径。根据剂量和给药途径,优选的剂量和/或有效量的给药次数可能因受试者反应而异。在任何情况下,负责给药的从业者将确定组合物中活性成分的浓度和个体受试者的适宜剂量。
在某些实施方案中,药物组合物可包含,例如,至少约0.1%的活性化合物。在其它实施方案中,活性化合物可占例如约2%至约75%之间或约25%至约60%之间的重量单位,和其中可推导的任何范围。当然,每一个治疗有用组合物中的活性化合物的量可以以任何给定单位剂量的化合物获得适当剂量的方式制备。制备这类药物制剂的领域的熟练技术人员将考虑一些因素,例如溶解性、生物利用度、生物半衰期、给药途径、产品保质期以及其它药理学考虑,因此,各种剂量和治疗方案可能是需要的。
在其它非-限制性实例中,剂量也可包含每次给予从约1微克/kg/体重、约5微克/kg/体重、约10微克/kg/体重、约50微克/kg/体重、约100微克/kg/体重、约200微克/kg/体重、约350微克/kg/体重、约500微克/kg/体重、约1毫克/kg/体重、约5毫克/kg/体重、约10毫克/kg/体重、约50毫克/kg/体重、约100毫克/kg/体重、约200毫克/kg/体重、约350毫克/kg/体重、约500毫克/kg/体重,至约1000mg/kg/体重或更多,和其中可推导的任何范围。在从此处列出的数字可推导的范围的非限制性实例中,可根据上述数字给予约5mg/kg/体重-约100mg/kg/体重、约5微克/kg/体重-约500毫克/kg/体重等的范围。
vii.定义
术语“aa”指氨基酸。氨基酸替代由分子中使用字母代码的氨基酸位置(例如303)表示(数字前面的字母表示被替换的氨基酸,而数字后面的字母表示被引入的氨基酸)。
如本文所用的术语“部分”,当提及蛋白(如在“给定蛋白的部分”中)指该蛋白的片段。片段的大小范围可为从4个氨基酸残基到整个氨基酸序列减去1个氨基酸。
如本文所用的术语“蛋白”和“多肽”指包含通过肽键连接的氨基酸的化合物,并可互换使用。
如本文所用的,术语“融合蛋白”指含有与外源蛋白片段(由非-精氨酸酶蛋白组成的融合配偶体)连接(或可操作地连接)的感兴趣的蛋白(即人精氨酸酶或其变体)的嵌合蛋白。融合配偶体可以提高血清半衰期、溶解度,或者两者。它还可以提供亲和标记(例如,his-标签),以便从宿主细胞或培养上清液或两者中纯化重组融合蛋白。
术语“在可操作的组合中”、“按可操作的顺序”和“可操作地连接”指的是核酸序列以产生能够指导给定基因的转录和/或所需蛋白分子的合成的核酸分子的方式连接。该术语还指以产生功能蛋白的方式连接氨基酸序列。
如本文所用的术语“km”指酶的米-曼二氏常数(michaelis-mentenconstant),并被定义为特定底物的浓度,在此浓度下,给定的酶在酶催化反应中达到其最大速率的一半。
如本文所用的术语kcat指每单位时间每个酶位点将底物分子转化为产物并且其中酶以最大效率工作的底物分子的周转数或数量。
如本文所用的术语kcat/km是专一性常数,它是酶将底物转化为产物的效率的量度。
术语“mn-hargi”指具有mn(ii)辅因子的人i型精氨酸酶。术语“co-hargi”指具有co(ii)辅因子的人i型精氨酸酶(突变或天然的)。
术语“ic50”是半数最大(50%)抑制浓度(ic),因而是有效性的量度。
术语“聚乙二醇化”指与聚乙二醇(peg)的缀合,聚乙二醇由于其高度的生物相容性和易于修饰,已被广泛用作药物载体(harrisetal.,2001)。对各种药物、蛋白和脂质体的连接已显示改善停留时间和降低毒性(greenwaldetal.,2000;zalipskyetal.,1997)。peg可通过链末端的羟基和经由其它化学方法偶联(如共价连接)于活性剂;然而,peg本身每个分子最多只能限定两个活化剂。在不同的方法中,peg和氨基酸的共聚物已作为新的生物材料而被研究,它保留了peg的生物相容性性质,但也具有每分子许多连接点的额外优点(提供更大的药物负荷),并且它可经综合设计,以适应各种应用(nathanetal.,1992;nathanetal.,1993)。
术语“基因”指包含生产多肽或其前体所需的控制和编码序列的dna序列。只要保留所需的酶活性,就可以由全长编码序列或由编码序列的任何部分编码多肽。
术语“受试者”指动物,包括人。
术语“野生型”指具有从天然存在的来源分离的基因或基因产物特征的基因或基因产物。野生型基因是群体中最常见的基因,因此被任意指定为该基因的“正常”或“野生型”形式。相比之下,术语“修饰的”或“变体”或“突变体”是指与野生型基因或基因产物相比,在序列和/或功能性质上表现出改变(即改变的特性)的基因或基因产物。注意到可分离天然存在的突变体;它们由当与野生型基因或基因产物相比时它们具有改变的特性这一事实来确定。
viii.药剂盒
本发明提供药剂盒,例如治疗性药剂盒。例如,药剂盒可包括一种或多种如本文描述的药物组合物和任选的它们的用法说明。药剂盒还可包括用于实现给予此类组合物的一个或多个装置。例如,本药剂盒可以包括药物组合物和用于实施将该组合物直接经静脉内注射到癌性肿瘤的导管。在其它实施方案中,本药剂盒可包括稳定的多聚精氨酸酶或分离的聚乙二醇化精氨酸酶的预填充的安瓿,其任选地配制为药物,或冻干,与递送装置一起使用。
药剂盒可包含带有标签的容器。合适的容器包括,例如,玻瓶、小瓶和试管。容器可由各种材料例如玻璃或塑料形成。容器可容纳包含抗体的组合物,所述抗体对于治疗性或非-治疗性应用是有效的,如上文描述的。容器上的标签可指明组合物用于特定疗法或非-治疗性应用,标签还可指明体内或体外使用的指导,例如上文描述的那些。本发明的药剂盒通常包含上文描述的容器和一个或多个其它容器,这些容器包含从商业和用户的角度所需的材料,包括缓冲剂、稀释剂、填充剂、针头、注射器和带有使用说明的包装插页。
ix.实施例
以下实施例用来说明本发明的某些优选的实施方案和方面,且不应被解释为对其范围的限制。在以下实验的公开内容中,应用了以下缩写:eq(当量);m(摩尔的);µm(微摩尔的);mm(毫摩尔的);n(正常的);mol(摩尔);mmol(毫摩尔);µmol(微摩尔);nmol(纳摩尔);g(克);mg(毫克);µg(微克);l(升);ml(毫升);µl(微升);cm(厘米);mm(毫米);µm(微米);nm(纳米);ec(摄氏度);mw(分子量);pbs(磷酸盐缓冲盐水);min(分钟)。
实施例1
掺入和测定i型精氨酸酶中的金属含量
mn2+和co2+的掺入可通过纯化精氨酸酶,接着与10mm金属于50℃10分钟的孵育步骤实现。为测定精氨酸酶制剂的最终金属含量和特性,mn-hargi(145µm)、co-hargi(182µm)的蛋白样品和相关透析缓冲液(100mmhepes,ph7.4)在2%硝酸中稀释并通过电感耦合等离子体质谱(icp-ms,departmentofgeologicalsciences,universityoftexasataustin)分析,以通过从最终蛋白样品的金属浓度减去在透析缓冲液中发现的金属浓度并除以蛋白浓度,对蛋白的钴、铁、锰和锌含量定量。为测定蛋白浓度,根据氨基酸序列计算hargi的消光系数(gill和vonhippel,1989)。在6m胍鎓盐酸盐、20mm磷酸盐缓冲液(ph6.5)的最终缓冲液浓度下,根据计算的ε280=24,180m-1cm-1计算i型精氨酸酶的所有蛋白浓度。为了比较,还使用bsa的稀释液作为标准,通过bca测定法,计算了精氨酸酶浓度。使用这种方法,发现与co2+一起孵育的精氨酸酶样品含有2.1±0.5当量co和0.4±0.1当量fe,不含可检测的量的zn或mn。与mn2+一起孵育的样品含有1.5±0.2当量mn和0.4±0.1当量fe,且无可检测的量的zn或co。因此,热孵育是一种有效的钴掺入方法。
对钴负载的其它研究已表明,较高比例的钴负载是可以达到的,并且产生更高的比活性。这些研究的结果示于下表和图3中。
表2
co-i型精氨酸酶钴负载
*绘图
标注星号的数据示于图3中。
实施例2
co-arg及其变体对肝细胞癌细胞和转移性黑素瘤的细胞毒性
为测试工程改造的精氨酸酶的体外细胞毒性,使各种浓度(0-100nm)的mn-argi、co-argi或co-hargi变体与hcc(hep3b)细胞或黑素瘤(a375)细胞(美国典型培养物保藏中心)在96-孔板中,以500个细胞/孔的接种密度,在补充有胎牛血清的dmem培养基中一起孵育。于37℃孵育24小时后,用含不同浓度的精氨酸酶的培养基按一式三份处理细胞。对照溶液是在培养基中的平衡盐溶液。于37℃和5%co2下维持经处理的细胞。在第1、3、5和7天通过加入100µl/孔的mtt(5mg/ml),和在每小时1-2次温和的搅动下孵育4小时,用标准mtt测定法(sigma-aldrich)测试细胞。此后,吸出溶液,然后在每孔中加入200µldmso。使用自动平板阅读器对每孔解析570nm处的吸光度,以确定与对照相比幸存细胞的相对数量。使得到的数据对指数方程进行拟合,以确定精氨酸酶细胞毒性的表观ic50值。计算来自第5天的ic50值,得到mn-hargi的5±0.3nm(~0.18µg/ml)的ic50值和co-hargi的0.33±0.02nm(~0.012µg/ml)的值。因此,针对hcc,co-argi酶似乎是mn替代的酶的细胞毒性的15倍。针对转移性黑素瘤细胞系(a375),mn-hargi导致4.1±0.1nm(~0.15µg/ml)的表观ic50。用co-hargi孵育导致细胞毒性的13倍增加,其表观ic50为0.32±0.06nm(~0.012µg/ml)。
实施例3
工程改造fc-精氨酸酶融合蛋白以提高体内半衰期
对iggfc结构域的融合已被广泛应用于延长治疗多肽例如tnf-α抑制剂依那西普(enbril™)的体内半衰期。fc结构域结合于fcγrn受体,后者在血管内皮和许多其它组织上表达(roopenian和akilesh,2007)。fcγrn对iggfc结构域的亲和力是强烈ph依赖性的。结合发生在内体隔室的酸性ph值下,允许蛋白再循环到细胞表面,从而规避蛋白水解的降解。在细胞表面,fc结构域从fcγrn释出,因为结合亲和力在生理ph下是非常低的。据估计,经由fcγrn的内体再循环可使人中免疫球蛋白的血清半衰期增加至少4-7倍,至7-14天左右。fc融合物利用这一特性,以赋予短寿命分子长的半衰期。然而,人精氨酸酶是同型三聚体,因此如果与iggfc(其本身是一种二聚体)融合,得到的fc-精氨酸酶多肽将可能形成高分子量聚集体。
这个问题通过使用突变体形式的精氨酸酶来避免,突变体形式的精氨酸酶破坏三聚作用并在单体形式上是稳定的。i型精氨酸酶的三聚作用和亚单位界面已经做了一些详细的研究(lavuloetal.,2001)。已表明glu256gln的单个氨基酸替代破坏三聚作用,导致单体i型精氨酸酶的形成(sabioetal.,2001)。这种变体的表达和纯化后,稳态动力学分析表明与1,320s-1mm-1的kcat/km的co-hargi相比几乎相同的活性。
然后将这个构建体克隆到fc表达载体。fc表达载体是基于ptrc99a质粒(amersham)的构建体,其含有dsba前导序列,接着是iggfc编码区、ecori限制性内切位点和终止密码子。单体精氨酸酶基因通过用ecori消化载体和基因二者而被置于fc编码区后面的框架中,随后连接并将其转化至大肠杆菌(bl21)用于测序和表达。因为iggfc通常是一种糖基化蛋白,重组igg或fc融合物的表达到目前为止已经在重组哺乳动物细胞中进行,与细菌不同,重组哺乳动物细胞能够进行n-连接的糖基化。然而,虽然asn297的糖基化对与活化和抑制性fcγ受体(在人中为fcγri-iii)的结合至关重要,但它对与fcγrn的亲和力或ph依赖性结合没有明显的影响(tao和morrison,1989;simmonsetal.,2002)。因此,在细菌中表达的无糖基化igg抗体在灵长类动物中显示出与哺乳动物细胞中表达的完全糖基化抗体几乎不能分辨的血清持久性(simmonsetal.,2002)。与早期流行的观念相反,igg抗体和fc蛋白可在发酵罐的大肠杆菌中有效地表达至g/l水平。从技术上讲,大肠杆菌表达更简单且更快。此外,由于所产生的蛋白是无糖基化的,它不显示聚糖异质性,这是治疗性糖蛋白表达中的一个重要问题(jefferis,2007)。通过蛋白a色谱纯化融合蛋白,通过fplc凝胶过滤色谱法定量正确折叠的、二聚体fc-精氨酸酶融合物相对于未二聚化多肽的产率。这种制剂导致了适合于体内试验的高度活性和非常稳定形式的人精氨酸酶。
实施例4
精氨酸酶的聚乙二醇化
纯化精氨酸酶,然后用cocl2制成10mm并于50℃下加热10分钟。在离心去除任何沉淀后,使用100,000mwco过滤设备(amicon),对peg-5000精氨酸酶进行广泛的缓冲液交换(含10%甘油的pbs),并用0.2微米注射器过滤器(vwr)除菌。使用limulusamebocytelysate(lal)试剂盒(capecodincorporated)分析所有聚乙二醇化酶的脂多糖(lps)含量。
发现聚乙二醇化co-hargi与野生型酶具有几乎相同的血清稳定性并显示出1690±290s-1mm-1的kcat/km值。
实施例5
l-arg在小鼠模型中的血清耗竭
通过单次ip注射500µg药学制备的聚乙二醇化co-hargi或等量的pbs处理balb/c小鼠。在0、48、72和96小时的时间点经心脏静脉穿刺处死小鼠以收集血液。将血样立即与400mm柠檬酸钠缓冲液(ph4)按50:50(v/v)混合,允许凝结30分钟并离心使血清分离。然后将得到的血清在10,000mwco装置(amicon)上过滤以除去大蛋白和沉淀物,收集流过液以供分析。l-精氨酸标准品、对照小鼠血清和实验样品用opa(agilent)导出,在c18反相hplc柱(agilent)(5µm,4.6x150mm)上分离,分离操作基本上如agilenttechnologies(publicationnumber:5980-3088)所述,除了通过将流速降低1/2和使采集时间加倍以获得更好的峰分离,对分离方案稍作修改之外。通过绘制l-arg峰面积对比浓度的关系构建l-精氨酸标准曲线,以定量血清l-arg水平。单剂量的药学制备的co-hargi足以保持l-arg在或低于检出限超过3天(图1)。
实施例6
用co-hargi治疗hcc肿瘤异种移植物
对裸小鼠侧腹皮下注射从75%汇合组织培养物中收集的~106个hcc细胞。在hcc异种移植的肿瘤生长至直径~0.5cm3(第9天)后,将小鼠分为两组。实验组在第9天和第12天接受500µg药学优化的co-hargi的ip注射。对照组在第9天和第12天接受pbs的ip注射。如可在图2中所见到的,pbs处理的肿瘤的尺寸在第15天增加了3-倍。形成鲜明对比的是,co-hargi治疗的肿瘤的尺寸在第15天减少。mn-hargi治疗的肿瘤仅在生长速度上表现出迟滞(chengetal.,2007)。co-hargi在体外和体内似乎都是一种针对hcc的高效的化疗剂。
实施例7
用co-hargi和抗-pd-l1ab破坏肿瘤微环境中的l-精氨酸平衡
人i型精氨酸酶(hargi)是一种mn2+-依赖性酶,其在血清中表现出低活性和低稳定性。骨髓-衍生的抑制细胞(mdsc)表达hargi和一氧化氮合酶(nos),其控制肿瘤微环境中l-精氨酸的可用性,进而调节t-细胞的功能。用mdsc耗竭l-精氨酸与t-细胞抗-肿瘤功能受损和肿瘤逃避宿主免疫有关。对在外周血单核细胞、骨髓单核细胞和cd34+细胞中l-精氨酸生物合成途径的酶的表达进行分析,揭示这些细胞表达低水平的otc和ass,提示这些细胞的生理功能对外源/胞外l-精氨酸的依赖性。基于这一发现,考虑到长期消耗l-精氨酸可能会对mdsc群产生负面影响,从而增强肿瘤生长的免疫调节。这一假说使用通过用co2+替代mn2+天然辅因子而开发的工程化hargi(aeb1102)检验,与内源性hargi相比,其导致催化活性和血清稳定性明显提高。工程化酶也如上所述被聚乙二醇化。
单独和与抗-pd-l1和抗-pd-1单克隆抗体(mab)联合给药的鼠ct26结肠癌模型中慢性的广泛聚乙二醇化co-hargi-介导的体内l-精氨酸耗尽的影响被测试。
雌性envigobalb/c小鼠(balb/cannhsd)被用于这些研究。它们在测试的第一天为6-7周龄。在第0天用5.0e+05个ct26.wt细胞皮下植入试验动物。所有小鼠均分至研究组,并如下开始治疗:
aeb-001-1037
-组1:媒介(pbs)ip,q7dx4,加同种型对照,10mg/kgip,(q3dx2;3off)x4
-组2:aeb1102,(3mg/kgip,q7dx4)
-组3:抗-pd-l1ab,10mg/kgip,(q3dx2;3off)x4
-组4:aeb1102,3mg/kgip,q7dx4;加抗-pd-l1ab,10mg/kgip,(q3dx2;3off)x4;
aeb1102给予4次,每周一次,(第3、10、17、24天)
抗-pd-l1给予8次,每周隔两天一次,隔3天一次(第3、6、10、13、17、20、24、27天)
每日观察所有动物的临床体征至少一次。每周记录个体体重和肿瘤体积3次。各小鼠在肿瘤大小达到2000mm3时终止。
用aeb1102(聚乙二醇化co-hargi)体内治疗ct26小鼠导致与靶向pd-1和pd-l1的标准免疫调节抗体可比较的疗效。重要的是,与单用aeb1102和单用免疫疗法比较,aeb1102与抗-pd-1和pd-l1mab的组合疗法导致明显的抗-肿瘤协同效应或至少相加的抗-肿瘤效应。
来自本研究的数据图表显示于图4中。数据反映了用聚乙二醇化co-hargi与抗-免疫检查点蛋白受体(抗-pd-1)和配体(抗-pd-l1)联合治疗的效果。在图中,上面的曲线是同种型对照抗体,第二条曲线是抗-pd-l1抗体,第三条曲线是聚乙二醇化co-hargi,而最下面的曲线是聚乙二醇化co-hargi和抗-pd-l1抗体的组合治疗。如数据所示,2种药物的组合对肿瘤生长的抑制具有大于相加的效应。
对淋巴细胞t细胞激活的作用也在第3天采集的样品中进行了测定。表3显示了4个组中表达cd45+的总的活细胞的百分比,以及还是cd8+的cd45+细胞的百分比。这些数据也在图6和7中以图形形式显示。
表报告了均数±sem
总体来说,这些结果表明,破坏肿瘤微环境中的l-精氨酸生理平衡抑制肿瘤生长,并且进一步使肿瘤对免疫治疗敏感。
实施例8
用argi和ox40治疗结肠癌的效果
将mc38结肠癌细胞的悬浮液注入雌性c57bl/6小鼠的侧腹。当肿瘤体积在第0天达到75-100mm3时,将小鼠随机分组。使用测径器一周两次测量肿瘤体积。在第0天开始治疗。
每两周对第一组注射10mg/kg同种型对照,持续6周,第二组每周注射3mg/kgco-i型精氨酸酶,持续6周,第三组每周注射10mg/kg抗-ox40ab,持续6周,和第四组每周注射3mg/kgco-argi和10mg/kg抗ox40ab,持续6周。来自本研究的数据示于图5中。虽然co-argi似乎是在次优剂量下给予的,但发现一种趋势,其中argi和aox40的组合对肿瘤体积的减少或抑制具有大于相加的效应。
本文公开和要求保护的所有组合物和方法都可以根据本公开内容制作和执行,而不需要进行过度实验。虽然本发明的组合物和方法已经根据优选的实施方案进行了描述,但对于本领域的技术人员来说,在不背离本发明的概念、精神和范围的情况下,显然可以对所述组合物和方法以及在本文描述的方法的步骤或步骤顺序中应用各种变化。更特别地,显而易见,在化学和生理学上均相关的某些药剂可以替代本文所述的药剂,同时会取得相同或类似的结果。对于本领域技术人员而言显而易见的所有这样的类似替代和修饰被视为在由所附权利要求书定义的本发明的精神、范围和概念内。
参考文献
以下参考资料,在它们提供了对本文所述内容进行补充的示例性程序或其它细节方面,在此通过引用特别地并入本文。
camaetal.,biochemistry,42:7748-7758,2003.
chengetal.,cancerlett.,224:67-80,2005.
chengetal.,cancerres.,67:309,2007.
chengetal.,cancerres.,67:4869-4877,2007.
dillonetal.,med.sci.monit.,8:br248-253,2002.
dowlingetal.,cellmol.life.sci.,65(13):2039-55,2008.
duranteetal.,clin.exp.pharmacol.physiol.,34:906-911,2007.
ensoretal.,cancerres.,62:5443-5450,2002.
feunetal.,j.neurooncol.,82:177-181,2007.
gill和vonhippel,anal.biochem.,182:319-326,1989.
greenwaldetal.,crit.revtherapdrugcarriersyst.,17:101-161,2000.
harkkietal.,biotechnology,7:596-603,1989.
harrisetal.,clin.pharmacokinet.,40(7):539-51,2001.
hopwoodetal.,in:geneticmanipulationofstreptomyces,alaboratorymanual,johninnesfoundation,norwich,conn.,1985.
izzoetal.,j.clin.oncol.,22:1815-1822,2004.
jefferis,expertopin.biol.ther.,7:1401-1413,2007.
lavuloetal.,j.biol.chem.,276:14242-14248,2001.
lopezetal.,febsj.,272:4540-4548,2005.
lordanescu,j.bacteriol,12:597601,1975.
melloretal.,gene,24:1-14,1983.
nathanetal.,bioconjchem.,4:54-62,1993.
nathanetal.,macromolecules,25:4476-4484,1992.
pardoll,drew,naturereviewscancer12,252-264(april2012
penttilaetal.,gene,61:155-164,1987.
remington’spharmaceuticalsciences,第18版.mackprintingcompany,1289-1329,1990.
roopenian和akilesh,nat.rev.immunol.,7:715-725,2007.
sabioetal.,febslett.,501:161-165,2001.
santhanametal.,circ.res.,101:692-702,2007.
savocaetal.,cancerbiochem.biophys.,7:261-268,1984.
scottetal.,br.j.cancer,83:800-810,2000.
shenetal.,cancerlett.,231:30-35,2006.
sibakovetal.,eur.j.biochem.,145:567572,1984.
simmonsetal.,j.immunol.methods,263:133-147,2002.
taoetal.,j.immunol.,143:2595-2601,1989.
ward,embo-alkoworkshoponmolecularbiologyoffilamentousfungi,helsinki,119-128,1989.
wheatley和campbell,pathol.oncol.res.,8:18-25,2002.
yoonetal.,int.j.cancer,120:897-905,2007.
zalipskyetal.,bioconjugchem.,8:111-118,1997.
- 还没有人留言评论。精彩留言会获得点赞!