紫芝液体深层发酵菌丝体均一多糖及其制备方法以及应用与流程
本发明涉及紫芝液体深层发酵菌丝体均一多糖GS-A-1、其制备方法及其在制备抗肿瘤药物及免疫调节药中的用途。
背景技术:
肿瘤是当今世界直接危及人类生命的一种最常见、最严重的疾病,因而如何有效地预防和治疗肿瘤已成为医学领域研究的一个热点。传统的手术、放化疗方法在延长肿瘤患者生命的同时会产生内脏器官损伤、免疫功能抑制等副作用,使病人的生活质量明显下降。目前,真菌多糖因具有毒副作用小、安全性高、抑制肿瘤效果好等优点,常用于肿瘤的辅助治疗,其作为一种免疫增强剂辅助放疗、化疗,已成为肿瘤治疗的重要手段。食药用真菌具有抗肿瘤抗病毒、降血脂、延缓衰老、护肝排毒、促进核酸和蛋白质生物合成等多种功效,在中国有悠久的使用历史。国内外大量研究表明,真菌多糖是从真菌子实体、菌丝体、发酵液中分离得到的,为真菌类药物的药效部位,是能够控制细胞分裂分化,调节细胞生长衰老的一类活性多糖。紫芝(Ganodermasinense)作为灵芝属真菌(2010版中国药典),其有较明显的抗肿瘤、提高免疫力的功效。由于其制备工艺不明确,多糖含量较低(一般<60%重量)。另外紫芝粗多糖组分复杂,主要药效部位和组分尚未阐明。此外由于紫芝子实体资源稀少,市场上少见紫芝产品。紫芝粗多糖溶解度很差,粘度较大,颜色深,味重,成分复杂,分子量从几千到上千万,难找到合适的质量控制方法,而且活性成分不明确,构效关系和作用机制也无从考察。若能将粗多糖分离纯化从中得到活性均一多糖,不仅能提高药效水平,并且对质量控制、构效关系和作用机制考察有很大的帮助。中国专利200710045369.4“一种紫芝发酵方法及制得的紫芝菌丝体”公开了一种紫芝液体深层发酵产生菌丝体的方法,该菌丝体用水提醇沉法提取得到的粗多糖含量在39~75%重量,其体内药理实验中,口服剂量为40mg/kg/天时有一定抗肿瘤活性。中国专利200710045368.x“紫芝菌丝体胞内多糖及其制备方法和应用”公开了一种从紫芝液体深层发酵产生的菌丝体中提取制备得到粗多糖的方法。该方法涉及的水提醇沉法提取得到粗多糖含量在39~75%重量。其药理实验的剂量也为40mg/kg/天(给药方式:口服喂药)。杨国红,杨义芳,金隽迪(紫芝液体深层发酵的抗肿瘤活性部位研究[J].中草药,2008,39(6):877-880)对紫芝液体深层发酵的抗肿瘤活性多糖进行了研究,从紫芝菌丝体中提取得到胞内多糖,其药理实验的剂量也为40mg/kg/天(给药方式:口服喂药)。然而,以上专利申请和文章均未涉及具抗肿瘤活性的均一多糖的研究,而且粗多糖存在着多糖含量不高、给药剂量不优、活性成分不够明确和抗肿瘤作用机制无从考察等不足。寻找多糖含量高、给药剂量更优、活性成分明确单一的的抗肿瘤均一多糖无论对作用机制研究,还是在抗肿瘤药物中应用都具有重要意义。
技术实现要素:
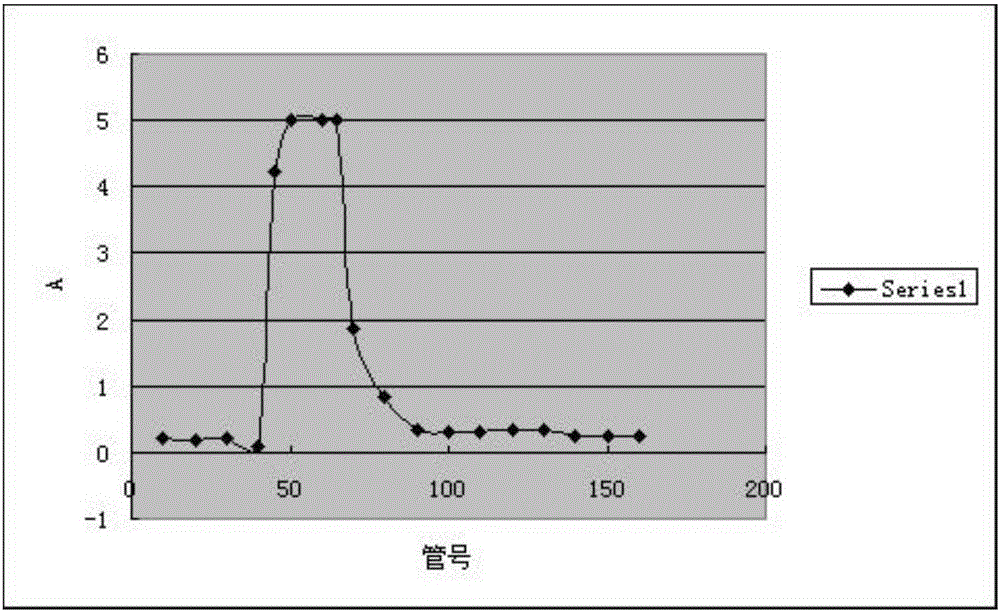
本发明要解决的技术问题是提供一种多糖含量高、可在制备抗肿瘤药物与免疫调节药物中应用的紫芝液体深层发酵菌丝体抗肿瘤均一多糖。如本发明所使用,“液体深层发酵”是发酵工业上常用的一种发酵方法,它的培养基是液态的。如本发明所使用,“菌丝体”是菌丝集合在一起构成一定的宏观结构。如本发明所使用,“均一多糖”是单糖通过聚合而成的多糖,聚合的糖有特定的结构单元。均一多糖可分为相同单糖形成的聚糖和不同单糖形成的杂多糖。本发明的第一个目的在于提供一种结构全新的紫芝液体深层发酵菌丝体均一多糖,将其命名为GS-A-1,其特征在于,所述均一多糖的总多糖含量为大于95%重量,优选为大于98.5%重量;且所述均一多糖GS-A-1的分子量为10000-30000道尔顿,优选为21190道尔顿。根据本发明的一个优选的实施方式,经TLC(薄层色谱分析)和HPLC(高效液相色谱法)分析可以得知,所述均一多糖GS-A-1中的多糖主要由半乳糖和甘露糖组成,所述半乳糖、甘露糖的摩尔比为1.2-2.2∶1,优选1.7∶1。进一步,经高碘酸氧化及Smith降解分析得知,所述均一多糖GS-A-1是由半乳糖和甘露糖通过(1→2),(1→6)连接的杂聚多糖。本发明的第二个目的在于提供上述紫芝液体深层发酵菌丝体均一多糖GS-A-1的制备方法,其特征在于,包括如下步骤:紫芝液体深层发酵菌丝体经水提醇沉得到粗多糖,粗多糖经超滤、浓缩和柱层析,得到紫芝液体深层菌丝体均一多糖GS-A-1。上述制备方法简单易行、生产周期短、成本较低廉。根据本发明的一个优选的实施方式,超滤步骤通过超滤膜进行。选择截留分子量为1-5万的超滤膜。更优选地,所述超滤膜的截留分子量为3万。根据本发明的一个优选的实施方式,所述超滤膜可由聚酰胺聚醚砜膜或醋酸纤维素制成。根据本发明的一个优选的实施方式,浓缩步骤通过纳滤膜进行。优选由聚酰胺复合材料构成的纳滤膜。优选截留分子量为150-300的纳滤膜。根据本发明的一个优选的实施方式,柱层析步骤通过加负压进行。采取的仪器设备为蠕动泵,自动收集仪。根据本发明的一个优选的实施方式,本发明所选择的填料为阴离子交换树脂。根据本发明的一个特别优选的实施方式,柱层析选择的填料阴离子交换树脂为DEAE-cellulose52。本发明制备均一多糖GS-A-1的具体步骤如下:紫芝菌丝体(本发明所述“紫芝菌丝体”根据中国专利200710045369.4“一种紫芝发酵方法及制得的紫芝菌丝体”中公开的方法所得)加水提取,提取液减压浓缩后,醇沉(通常使用乙醇),沉淀部分干燥至恒重后,再溶解于水,过滤,用一定分子量的超滤膜超滤,再用一定分子量的纳滤膜分别浓缩得到截留液和透过液,将上述透过液冷冻干燥,得到分子量小于3万的紫芝菌丝体多糖组分GS-A;再经过阴离子交换树脂柱得到紫芝液体深层发酵菌丝体均一多糖GS-A-1。本发明利用膜技术和柱层析将紫芝液体深层发酵菌丝体粗多糖纯化成均一多糖,药理实验表明纯化后均一多糖GS-A-1在抗肿瘤药物的应用中有明显的活性。本发明方法得到的紫芝液体深层发酵菌丝体均一多糖GS-A-1含量为大于95%重量,优选为98.5%重量(苯酚-硫酸法检测),其分子量为21190Da。将该均一多糖部位进行药理活性研究,结果表明该均一多糖与环磷酰胺(CTX)联用具有增效减毒及提高免疫力的作用。因此,本发明的第三个目的在于提供上述紫芝液体深层发酵菌丝体均一多糖GS-A-1在制备抗肿瘤药物和调节免疫力药物中的应用。与现有的技术相比,本方法提供的紫芝液体深层发酵菌丝体均一多糖GS-A-1的多糖含量更高、纯度更高,活性成分单一明确,给药剂量更优,且其制备工艺亦适于工业化生产。附图说明图1为本发明实施例1所得紫芝液体深层发酵菌丝体均一多糖GS-A-1的HPGPC图谱。图2为实施例1步骤3)中GS-A在DEAE中的洗脱曲线。图3为葡萄糖标准曲线。图4为GS-A-1水解后的TLC结果,其中1果糖、2岩藻糖、3阿拉伯糖、4甘露糖、5葡糖糖、6GS-A-1水解液、7木糖、8半乳糖、9鼠李糖、10核糖。图5A-5D分别为水、半乳糖、甘露糖、实施例1制得的GS-A-1的HPLC图谱。图6为GS-A-1的高碘酸标准曲线。图7A-7C分别为水、甘油、实施例1制得的GS-A-1降解样品的HPLC图谱。图8为实施例1制得的GS-A-1的紫外图谱。图9为实施例1制得的GS-A-1的红外图谱。图10为实施例1制得的GS-A-1的1H-NMR图谱。图11为实施例1制得的GS-A-1的13CNMR图谱。具体实施方式为了更好地理解本发明,通过以下实施例来说明,但这些实施例不构成对本发明的限制。实施例1:紫芝液体深层发酵菌丝体均一多糖GS-A-1的制备:1)粗多糖的制备:紫芝液体深层发酵菌丝体(所述紫芝液体深层发酵液通过中国专利200710045369.4“一种紫芝发酵方法及制得的紫芝菌丝体”中所述方法得到)具体方法如下:从山东购得的紫芝中分离得到紫芝菌种(Ganodermasinense),在无菌室中接种于经121℃消毒灭菌30分钟的平板培养基,28℃培养。4-6天后在无菌室中将菌种接种于经121℃消毒灭菌30分钟的摇瓶种子培养基,摇床上28±1℃震荡培养5-7天,挑选较好的菌种于发酵罐内培养5-6天,得到紫芝液体发酵菌丝体。将菌丝体用水在60~90℃提取1~3h,提取液减压浓缩,加入乙醇沉淀,沉淀部分干燥至恒重,得粗多糖。2)超滤分离:用200倍体积的水溶解粗多糖,过滤,取水溶液进行外压式超滤处理(超滤膜可以是由聚酰胺制成、截留分子量为3万的超滤膜;或由聚醚砜膜制成、截留分子量为1万的超滤膜;或由醋酸纤维素制成、截留分子量为5万的超滤膜),进样压力=0.2Mpa,浓溶液流出压力=0.15Mpa,将粗多糖水溶液分为截留液和透过液。将透过液用纳滤膜浓缩(纳滤膜是聚酰胺复合材料构成的,截留分子量为150、200或250),并冷冻干燥,得到透过液部分GS-A。3)柱层析分离:取2gGS-A,用少量去离子水溶液溶解,加入已平衡好的DEAE-cellulose52纤维素柱中,用去离子水洗脱,以10mL/min管收集洗脱液2个柱体积。每隔5~10管用苯酚-硫酸法测定其吸收值,并做洗脱曲线。洗脱曲线见附图2。分别将吸光度值较大的45,50,60,65号管的样品进行高效液相色谱分析,发现45号管样品表现为单峰,分别将45号管附近的样品进行高效液相色谱分析,发现42~47管的样品色谱图一致。合并42~47管的样品,浓缩,冷冻干燥。将该样品命名为GS-A-1。实施例2:苯酚硫酸法测定本发明紫芝液体深层发酵菌丝体均一多糖GS-A-1的多糖含量具体步骤如下:标准品配制:精密称定干燥的葡萄糖25mg,以水溶解并定容至100ml,配成250ug/ml标准品溶液,分别取0.05、0.1、0.15、0.2、0.25、0.3mL于试管中,加去离子水补足至0.3mL,分别加5%苯酚0.7mL充分摇匀,快速加入浓硫酸4mL,50℃水浴40min,冷却至室温,作为对照品。空白对照品配制:取0.6ml去离子水,加5%苯酚1.4ml,浓硫酸8ml,50℃水浴加热40min,冷却至室温,作为空白液。标准曲线绘制:将标准品溶液和空白对照品溶液用紫外分光光度法于488nm处测其吸收值,并做标准曲线。标准曲线见附图3。样品配制:取GS-A-1样品精密称定5mg,以去离子水溶解并定容至10ml,配制0.5mg/ml溶液,取0.05ml于试管中,加水补足至0.3ml,同标准品配制方法依次加入苯酚、浓硫酸,50℃水浴加热40min,冷却至室温,作为样品液。检测:将待测样品用紫外分光光度法于488nm处测其吸收值,用标准曲线测得GS-A-1的多糖含量为98.5%重量。实施例3:用葡聚糖标准品Dextron系列标定本发明紫芝液体深层发酵菌丝体均一多糖GS-A-1中多糖的分子量具体方法如下:色谱条件:TSK-GELGMPWxl色谱柱;流动相为水;流速:0.3mL/min;柱温35℃;检测器为:示差检测器。标品Dextron、GS-A-1样品分别用水溶解,进样。测定葡聚糖Dextron系列和GS-A-1的保留时间,以Dextron的保留时间-Dextron系列的分子量对数值做标准曲线,根据GS-A-1的保留时间以及标准曲线,可知GS-A-1峰位分子量为21190道尔顿。实施例4:紫芝液体深层发酵菌丝体均一多糖GS-A-1的结构解析1)完全酸水解:取5mg实施例1制得的GS-A-1,放入安瓿瓶中,加入2mL,2mol/L的三氟醋酸(TFA),充N2后封瓶,在110℃下水解4h。将瓶内溶液减压蒸干,然后加入甲醇1~2mL蒸干,重复三次操作,以完全除去TFA。再向样品中加入0.5mL的水使样品溶解。1.1GS-A-1水解后的TLC结果:在高效硅胶板(型号:HSGF254)上分别点多糖水解样品及单糖,分别以丙酮-水(24∶1);正丁醇-乙酸乙酯-异丙醇-醋酸-水-吡啶(35∶100∶60∶35∶30∶35)上行二次展开,取出晾干,用苯胺-邻苯二甲酸于110℃加热10min。TLC显示样品对应处有半乳糖和甘露糖两个斑点,见附图4。从图4可以看出,本发明GS-A-1是由半乳糖、甘露糖组成的一种杂多糖。1.2HPLC检测多糖单糖组分色谱柱条件:液相色谱柱:Kromasil100-5NH2(250×4.6mm)检测器:示差折光检测器流动相:乙腈∶水=3∶1柱温箱:35℃检测器温度:35℃流速:1mL/minHPLC检测结果见附图5A-5D。根据标品半乳糖与甘露糖的摩尔量,结合多糖样品HPLC的峰面积比,可以推断多糖GS-A-1单糖中半乳糖∶甘露糖=1.7∶1(摩尔比)。可以进一步证明TLC点板中存在半乳糖和甘露糖单糖。2)高碘酸氧化:2.1高碘酸标准曲线:30mmol/L高碘酸液配制:精密称取高碘酸钠322.08mg以水溶解并定容于50mL容量瓶。标准曲线的绘制:取8个250mL容量瓶分别依次加入高碘酸钠溶液0.2,0.4,0.6,0.8,1,1.5,2mL,加去离子水定容。放置10min后,以水作为空白,用分光光度法,在223nm的波长处测吸收度,以高碘酸钠浓度(μmol/L)为横坐标,光密度值为纵坐标作标准曲线。见图6。2.2高碘酸氧化反应:精密称取GS-A-110mg于10mL容量瓶中,然后加入15mmol/LNaIO4定容至刻度。放置在暗处反应(4℃),间隔时间(0h、2h、24h、48h、72h......)取样0.1mL,用蒸馏水稀释250倍,用紫外分光光度计,以蒸馏水作空白对照,在223nm波长处测吸光度值,直到吸光度值恒定为止。加乙二醇终止反应(中和剩余的高碘酸),高碘酸氧化完成。2.3用NaOH滴定甲酸的生成量:酚酞指示剂配制:称取1g酚酞,加95%乙醇100ml溶解均匀,即得。0.1mol/LNaOH滴定液标定:精密称取105℃恒重的邻苯二甲酸氢钾0.6g溶于50mL新沸过的冷水,酚酞作为指示液,滴定至溶液显粉红色。最终测得NaOH溶液浓度为0.0986mol/L。甲基红指示剂配制:精密称取0.1g甲基红固体,加0.05mol/LNaOH溶液7.4mL使之溶解,再加水稀释至200mL,即得。取2mL上述氧化液,加1滴甲基红作指示剂,用0.0986mol/LNaOH(用邻苯二甲酸氢钾标定)滴定至氧化液由红色变成黄色,计算得甲酸生成量。2.4结果显示:2.4.1吸光值恒定时测得223nm波长处A值为0.562。高碘酸液在24h后吸收值无变化。故可以得出高碘酸与样品反应的消耗量为0.127mol。2.4.2根据滴定结果最后计算得甲酸生成量为0.0493mmol。2.4.3而高碘酸消耗量高于甲酸生成量的二倍,说明存在被高碘酸氧化不产生甲酸的己糖残基1→2、1→2,6、1→4、1→4,6键型;有少量甲酸生成说明存在1→、1→6键型的己糖残基,高碘酸消耗量及甲酸生成量显示分子中的糖残基可能全部被氧化。3)Smith降解反应将乙二醇处理后的GS-A-1用高碘酸氧化后的溶液透析(Mw=3500),蒸馏水透析48小时。浓缩,加入NaBH4还原过夜。用冰醋酸中和至pH为6~7,透析,减压蒸至2mL左右,进行冷冻干燥。将还原的样品放入安瓿瓶中,加入2mL2mol/LTFA,充N2后封管,于110℃下水解4h,反应瓶中的溶液减压蒸干,再加入1~2mL的甲醇,蒸干,重复三次以除尽过量的TFA,水解后的样品用1mL水溶解,进行HPLC分析。3.1色谱条件:液相色谱柱:Kromasil100-5NH2(250×4.6mm)检测器:示差折光检测器流动相:乙腈∶水=3∶1柱温箱:35℃检测器温度:35℃流速:1mL/min3.2结果:见附图7A-7C。GS-A-1降解产物HPLC检测出大量甘油表明多糖GS-A-1含有可以产生甘油的键型,即1→、1→2、1→6、1→2,6,说明GS-A-1的糖苷键键型为1→、1→2、1→6、1→2,6。综上所述:GS-A-1的一级结构为:4)紫外图谱:见附图8。取GS-A-1样品进行紫外全波长扫描,在260nm和280nm处未见吸收,证明不含核酸和蛋白质。5)红外图谱:取1mgGS-A-1样品,用溴化钾压片进行IR检测。结果见附图9。GS-A-1红外光谱图有多糖特征吸收,3427.3cm-1的宽峰是-OH的伸缩振动。2924.8cm-1峰是糖的C-H伸缩振动,1640.1cm-1为糖水化物的特征峰,1400cm-1附近出现的峰是由于C-H的变角振动引起的。1147.5cm-1的峰是C-O的伸缩振动。GS-A-1在1074.2cm-1的峰是常见的吡喃糖环内酯和羟基的吸收产生的吸收峰,是由于糖环上C-O-C醚键的不对称伸缩振动。在874.7cm-1处有吸收峰,其可能为半乳糖的吸收峰,结构中含有α糖苷键。在799.5cm-1处有吸收峰,其可能为甘露糖的吸收峰,结构中含有α糖苷键。说明GS-A-1为吡喃糖,其糖苷键类型为α-构型。6)核磁图谱:核磁光谱(NMR):取多糖样品10mg装入核磁管,溶于重水中,在核磁共振仪上进行1H-NMR,13C-NMR等光谱分析。1H-NMR图谱分析:如图10,主要解决多糖结构中糖苷键构型问题,由于多糖的信号大多堆集在化学位移δ3.3~5.2的狭小范围内,δ3.3~4.3为糖环质子信号。除C1上质子信号易解析外其他C2~C6的质子信号相互重叠交叉,解析困难。从附图2中可见C1质子化学位移位于δ4.8~5.2间,则证明GS-A-1含α型吡喃己糖,与红外结果相符。13C-NMR图谱分析:如图11,均显示多重峰,表示多糖结构的复杂性。通过比较化学位移已归属的取代单糖或简单寡糖来归属未知多糖,将GS-A-1的13C-NMR图谱信号归属如下:δ104.3和δ100.6是糖的端基碳,这两个碳信号,说明该均一多糖由两个单糖组成,且它们的碳信号化学位移值均大于δ100,结合1H-NMR图谱,说明均一多糖GS-A-1糖苷键的构型为α-L型。δ63.8是未取代的C6,推测可能为不同连接方式的α-L-半乳糖与α-L-甘露糖的C6。实施例5:实施例1制得的紫芝液体深层发酵均一多糖GS-A-1的药理活性1)对H22肝癌昆明小鼠的增效减毒作用:将2只H22肝癌小鼠,处死后置于超净工作台,腹部去毛,剪开皮层,用无菌注射器抽取腹水,抽完后用0.5ml生理盐水再冲洗一次腹腔,合并腹水和洗液,稀释,显微镜下细胞计数,使肝癌细胞浓度成2×107个/ml。然后于昆明种小鼠右腋下,以0.2ml/只的量,接种肝癌细胞。将正常昆明种小鼠按照体重随机分组,分为正常对照组、模型组、环磷酰胺组、环磷酰胺与香菇多糖联合给药组、环磷酰胺与GS-A-1联合给药组。除正常对照组外,其它各组小鼠均以0.2ml/只的量接种腹水肺癌细胞于右腋下。从接种后第二天开始给药,连续给药7天。其中环磷酰胺以25mg/kg的剂量给药,多糖组均以1.5mg/kg的剂量给药。第9天给药后眼静脉取血,分离血清测血常规;处死后剥离瘤体,并取脾脏,称重。表1GS-A-1对昆明小鼠H22肝癌增效减毒作用*P<0.5,**P<0.01较CTX在昆明小鼠H22肝癌模型实验中,环磷酰胺(25mg/kg)和GS-A-1(1.5mg/kg)联合腹腔注射给药与环磷酰胺(CTX,25mg/kg)单独腹腔注射抑瘤率显著提高,脾重、白细胞和血小板均显著增加,香菇多糖组则无明显效果(见表1)。GS-A-1在昆明小鼠H22肝癌模型中对CTX具有增效减毒的作用。2)对Lewis肺癌C57BL小鼠的增效减毒作用:2只肺瘤小鼠,处死后置于超净工作台,腹部去毛,用75%酒精消毒剪开皮层,用无菌注射器抽取腹水,置离心管中,抽完后再用0.5ml生理盐水冲洗一次腹腔,合并腹水和洗液,加一定量的生理盐水混匀,离心,弃上清。用生理盐水洗2-3次,稀释混匀后细胞计数,调整肺癌细胞浓度为2×107个/ml,以0.2ml/只的量接种于小鼠右腋下。将正常昆明种小鼠按照体重随机分组,分为正常对照组、模型组、环磷酰胺组、环磷酰胺与香菇多糖联合给药组、环磷酰胺与GS-A-1联合给药组。环磷酰胺以50mg/kg的剂量,分别在第2,4,6天下午腹腔注射给药。多糖组以1.5mg/kg的剂量,从接种肿瘤细胞后第二天开始腹腔注射给药,连续12天。第14天眼静脉取血,测血常规;立即处死剥离瘤体、并取脾脏,称重。表2GS-A-1对C57BL小鼠Lewis肺癌增效减毒作用*P<0.01较CTX在C57BL小鼠Lewis肺癌模型试验中,环磷酰胺(25mg/kg)和GS-A-1(15mg/kg)联合腹腔注射给药与环磷酰胺(25mg/kg)单独腹腔注射相比,抑瘤率、脾重、白细胞和血小板指数均显著提高,而香菇多糖组效果则不明显(见表2)。GS-A-1在C57BL小鼠Lewis肺癌模型中对CTX具有增效减毒的作用。3)GS-A-1提高免疫力活性考察:紫芝多糖对小鼠淋巴细胞增殖能力的影响:采用H22肝癌小鼠,无菌条件下抽取腹水,调整细胞数为2×107个/ml,以0.2ml/只在昆明种小鼠腋下接种。小鼠接种后按照体重随机分组,分为模型组、环磷酰胺组、环磷酰胺与香菇多糖联合给药组、环磷酰胺与GS-A-1联合给药组。连续给药9d,其中环磷酰胺以25mg/kg的剂量给药,多糖组均以1.5mg/kg的剂量给药。于末次给药0.5h,取血后处死动物,无菌条件下剖取脾脏。放入100目不锈钢丝网内,以眼科剪剔除结缔组织和脂肪,置于盛有5-10ml冷D-Hank′s液的培养皿中,用针芯碾碎脾组织,制成细胞悬液,置于离心管中,1000r/min离心5min,弃上清。低渗处理去除RBC,再以冷D-Hank′s液洗涤细胞3次,加入10%NBS-RPMI-1640培养液1.5ml悬浮脾细胞,计数,调整细胞数为5×106个/ml后加入96孔板,每孔100ul,空白孔只加培养液,其他孔按分组设计加入ConA(50ugml-1)、LPS(25ugml-1)20ul,分别刺激T、B淋巴细胞。在CO2培养箱内培养48h后,加入10ulMTT,继续培养4h后吸弃所有上清,每孔加100ulDMSO终止反应,振荡器上振荡1min,酶标仪570nm读板,结果以OD值表示。紫芝多糖对H22肝癌小鼠NK细胞的影响:采用H22肝癌小鼠,无菌条件下抽取腹水,调整细胞数为2×107个/ml,以0.2ml/只的量在昆明种小鼠腋下接种。连续给药9d,于末次给药0.5h,取血后处死动物,无菌条件下剖取脾脏。放入100目不锈钢丝网内,以眼科剪剔除结缔组织和脂肪,置于盛有5-10ml冷D-Hank′s液的培养皿中,用针芯碾碎脾组织,使成细胞悬液,置于离心管中,1000r/min离心5min,弃上清。低渗处理去除RBC,再以冷D-Hank′s液洗涤细胞3次,加入10%NBS-RPMI-1640培养液1.5ml悬浮脾细胞,计数,调整细胞数为5×106个/ml后加入96孔板,每孔100ul,空白孔只加培养液,其他孔按分组设计加入ConA(50ug·ml-1)20ul,刺激T淋巴细胞。在CO2培养箱内培养48h后,加入10ulMTT,继续培养4h后吸弃所有上清,每孔加100ulDMSO终止反应,振荡器上振荡1min,酶标仪570nm读板,结果以OD值表示。调整靶细胞YAC-1细胞浓度为1×105个/ml,靶细胞自然释放孔加200ul靶细胞培养液,效应对照孔只加效应细胞培养液,反应孔加入靶细胞培养液100ul,刺激NK细胞。在CO2培养箱内培养48h后,加入10ulMTT,继续培养4h后吸弃所有上清,每孔加100ulDMSO终止反应,振荡器上振荡1min,酶标仪490nm读板,结果以OD值表示,并根据NK细胞杀伤率(%)=(实验孔OD值-效应细胞对照孔OD值)/靶细胞对照孔OD值×100%,计算NK细胞的杀伤率。表3GS-A-1提高免疫力活性考察*P<0.05,**P<0.01较CTX在昆明小鼠H22肝癌模型实验中,环磷酰胺(25mg/kg)和GS-A-1(1.5mg/kg)联合腹腔注射给药与环磷酰胺(25mg/kg)单独腹腔注射能显著增加B细胞,T细胞和NK细胞的数目与活力,与阳性对照香菇多糖相比,表现出更好的提高免疫力作用(见表3)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1