苯丙酸类衍生物的合成和用途的制作方法
本发明涉及药物化学领域,具体涉及新型苯丙酸类衍生物及其制备方法与在制药中的用途,尤其涉及苯丙酸类衍生物作为GPR120受体激动剂在预防和治疗糖尿病中的用途。
背景技术:
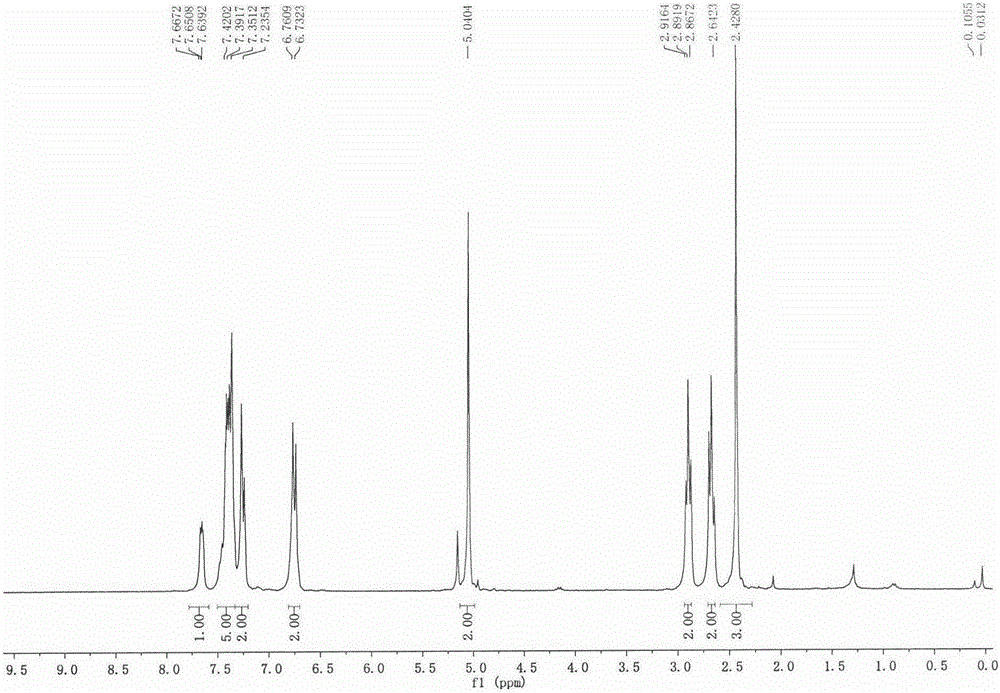
:糖尿病是一种糖代谢异常的代谢性疾病,临床上糖尿病主要可分为两种:I型和II型糖尿病。I型糖尿病也称胰岛素依赖型糖尿病,是一种慢性自身免疫性疾病,其特征是胰岛β细胞受损,导致胰岛素分泌不足。90-95%的糖尿病患者属于II型糖尿病。高血糖、胰岛素分泌受损和胰岛素抵抗是II型糖尿病的三个特征。持续的不受控制的高血糖会显著增加大血管和微血管并发症的风险,例如动脉粥样硬化、糖尿病肾病、糖尿病视网膜病变、糖尿病性心脏病和糖尿病神经病变等。GPR120,又称FFAR4,是长链脂肪酸的特异性受体。hGPR120编码基因定位于人类第10号染色体短臂(10q23.33)。hGPR120有两种亚型:长亚型和短亚型。其中短亚型由361个氨基酸构成,长亚型在氨基酸序列233-248处比短亚型多出16个氨基酸。通过RT-PCR检测,发现在人体15种组织中都有表达GPR120的mRNA,包括脑、垂体、心脏、肝、肾、肺、胃、肌肉、小肠、脾、皮下组织、结肠和胰腺等。其中,人与小鼠的小肠部位GPR120的mRNA表达最为丰富。同时,在人的四种不同的脂肪组织及味蕾细胞中,GPR120也有丰富的表达。在体外细胞系中,小鼠肠内分泌细胞系STC-1、ghrelin肿瘤细胞系MGN3-1、乳腺癌细胞系MCF-7及胰岛α细胞系αTC1-6均表达GPR120,但在NCI-H716、CaCo-2、T29、IEC-6、IEC-18及Min6细胞系中并无GPR120表达。此外,在人的成骨和破骨细胞中也有GPR120的表达。因此,GPR120可能与糖尿病、肥胖及骨代谢等疾病相关。GPR120受体被激活后可以促进GLP-1的释放。中、长链游离脂肪酸可以增加STC-1细胞内Ca2+的浓度,促进GLP-1的释放。采用GPR120siRNA下调GPR120的表达,可以显著降低中、长链游离脂肪酸对STC-1细胞内Ca2+的浓度的增加和GLP-1的释放。同时激活GPR120受体还可以增加胰岛素敏感性,改善胰岛素抵抗。在脂肪组织和巨噬细胞中,GPR120受体可以介导ω-3脂肪酸DHA和EPA的抗炎效应。因此激活GPR120受体可以发挥抗炎效应。此外有研究表明,GPR120功能的缺失可以导致人和鼠的肥胖。同时GPR120在肿瘤的生长和迁移、骨质疏松等疾病相关。综上所述,GPR120是糖尿病、肥胖等代谢性疾病的重要治疗靶点,其受体激动剂具有重要的开发价值。技术实现要素:本发明所要解决的技术问题在于设计、合成代谢稳定、具有较好成药性的新型GPR120激动剂。本发明公开了一系列苯丙酸衍生物或其药学上可接受的盐或溶剂化物,所述衍生物如通式I的化合物:其中R1为氟、氯、溴、碘、1~6个碳的直链或支链烷基;R2为芳环、取代芳环、芳杂环、取代芳杂环;R3为氢、氟、氯、溴、碘、1~6个碳的直链或支链烷基;X为碳、氧、硫、氮;n=0,1,2,3本发明通式I的化合物优选R1为氟、氯、溴、碘、甲基、乙基、正丙基、异丙基、正丁基、异丁基、环丙基、;其中,优选R2为苯环、单取代苯环、多取代苯环;优选R3为氢、氟、氯、溴、碘、甲基、乙基;优选X为碳、氧、硫、氮;优选n=0,1,2,3;本发明通式I的化合物更优选R1为氟、氯、溴、甲基;本发明通式I的化合物优选R2为苯环、单取代苯环、二取代苯环;本发明通式I的化合物更优选R3为氢、氟、氯;本发明通式I的化合物更优选X为碳、氧、氮;本发明通式I的化合物更优选n为1本发明优选的化合物如下:3-(3,5-二氟-4-((4-氟-4’-甲基-[1,1’-联苯基]-2-)甲氧基)苯基)丙酸I-1;3-(4-((4-氯-[1,1’-联苯基]-2-)甲氧基)-3,5-二氟苯基)丙酸I-2;3-(4-((4’-氯-4-氟-[1,1’-联苯基]-2-)甲氧基)-3,5-二氟苯基)丙酸I-3;3-(3,5-二氟-4-((4-氟-[1,1’-联苯基]-2-)甲氧基)苯基)丙酸I-4;3-(3,5-二氟-4-((4’-甲基-[1,1’-联苯基]-2-)甲氧基)苯基)丙酸I-5;3-(3,5-二氟-4-((4-氟-4’-甲氧基-[1,1’-联苯基]-2-)甲氧基)苯基)丙酸I-6;3-(3,5-二氟-4-((4-氟-3’-甲基-[1,1’-联苯]-2-)甲氧基)苯基)丙酸I-7;3-(4-([1,1’-联苯]-2-甲氧基)-3,5-二氟苯基)丙酸I-8;3-(4-((4,4’-二氯-[1,1’-联苯基]-2-)甲氧基)-3,5-二氟苯基)-丙酸I-9;3-(4-氯-4’-甲基-[1,1’-联苯基]-2-)-甲氧基)-3,5-二氟苯基)丙酸I-10;3-(3,5-二氟-4-((4-氟-2’-甲基-[1,1’-联苯基]-2-)甲氧基)苯基)丙酸I-11;3-(4-((4-氟-4’-甲基-[1,1’-联苯基]-2-)甲氧基)-3,5-二甲基苯基)丙酸I-12;3-(3,5-二溴-4-((4-氟-4’-甲基-[1,1’-联苯基]-2-)甲氧基)苯基)丙酸I-13;3-(3,5-二氯-4-((4-氟-4’-甲基-[1,1’-联苯]-2-)甲氧基)苯基)丙酸I-14;2-(3,5-二氟-4-((4-氟-4’-甲基-[1,1’-联苯基]-2-)甲氧基)苯氧基)乙酸I-15;2-(3,5-二氟-4-((4’-甲基-[1,1’-联苯基]-2-)甲氧基)苯氧基)乙酸I-16;本发明的另一目的是提供了一系列苯丙酸衍生物式I化合物的制备方法,如下反应式:具体包括下列步骤(1)式II化合物与中间体III通过Mitsunobo反应,得到化合物V,所采用的活化剂可以为DEAD、DIAD、ADDP、三丁基膦、三苯基膦等;溶剂选自苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙醚、乙酸乙酯、四氢呋喃、丙酮、乙腈、乙醇、甲醇、N,N-二甲基甲酰胺或二甲亚砜中的一种或多种的混合溶剂。反应温度为零下20℃至100℃,优选温度为0℃至60℃;(2)式II化合物在适合的条件下引入离去基团,得到化合物IV。离去基团可以为氯、溴、碘、对甲苯磺酰基、苯磺酰基、对硝基苯磺酰基、甲磺酰基等。(3)式IV化合物与式III化合物在碱的存在下发生亲核取代反应,得到式V化合物,所采用的溶剂选自苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、 乙醚、乙酸乙酯、四氢呋喃、丙酮、乙腈、乙醇、甲醇、N,N-二甲基甲酰胺或二甲亚砜中的一种或多种的混合溶剂,优选N,N-二甲基甲酰胺、四氢呋喃、乙腈或二氯甲烷;所采用的碱可以是三乙胺、二异丙基乙胺、吡啶、4-二甲氨基吡啶、1,8-二氮杂环[5,4,0]十一烯-7、碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾、叔丁醇钾、氢化钠、甲基氯化镁、异丙基氯化镁、叔丁基氯化镁、二异丙基胺基锂、六甲基二硅基胺基锂中的一种或多种混合使用;反应温度为零下20℃至100℃,优选温度为25℃至80℃;(4)式V化合物在碱性条件下水解得到式I化合物,所采用的碱可以是氢氧化锂、氢氧化钠、氢氧化钾、氢氧化铯、碳酸钾、碳酸钠、碳酸铯等;所采用的溶剂为水、四氢呋喃、甲醇、乙醇、乙腈、乙酸乙酯、丙酮、N,N-二甲基甲酰胺、二甲亚枫中的一种或多种混合溶剂。反应温度为零20℃至100℃,优选温度为25℃至80℃;在上述反应式中,R1、R2、R3如上述式I化合物中所定义;R4为烷基、芳基,包括甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、环丙基、环丁基、环戊基、环己基、苄基、对甲氧基苄基、苯基等。本发明的又一目的是提供了一系列苯丙酸衍生物式I化合物在制药中的用途。药效学实验结果表明,本发明式I化合物对GPR120具有较好的激动效果;体内代谢研究结果表明,本发明式I化合物能够表现出更好的代谢稳定性;更出人意料的是,式I化合物在羧基端苯环上引入二氟取代后,GPR120激动活性显著提高。上述实验结果提示,本发明化合物或其药学上可接受的盐可以用于制备抗糖尿病、肥胖等代谢性疾病。本发明还提供了一种预防或治疗糖尿病、肥胖等代谢性疾病的药物组合物,其中含有治疗有效量的式I化合物或其药学上可接受的盐和药学上可接受的载体。所述药物组合物可以是普通片剂或胶囊、缓释片剂或胶囊、控释片剂或胶囊、颗粒剂、散剂、糖浆剂、口服液、注射剂等制剂学上常规的制剂形式。附图说明图1化合物I-1的核磁共振氢谱;图2化合物I-5的核磁共振氢谱。具体实施方式下面通过实施例具体说明本发明的内容。在本发明中,以下所述的实施例是为了更好的阐述本发明,并不是用来限制本发明的范围。中间体的制备(4-氟-4’-甲基-[1,1’-联苯基]-2-)甲醇室温下,2-溴-5-氟苄醇(410mg,2mmol)、对甲基苯硼酸(299mg,2.2mmol)、无水碳酸钠(636mg,6mmol)溶于甲苯(10mL)、乙醇(5mL)、水(5mL)的混合溶液中。氮气保护下,加入四三苯基膦钯(116mg,0.1mmol),然后升至70℃搅拌过夜。原料消失后,反应液冷却至室温,硅藻土抽滤并用乙酸乙酯洗涤。滤液用乙酸乙酯萃取后用饱和食盐水洗涤,合并有机相并用无水硫酸钠干燥、脱溶并通过柱层析纯化,得黄色液体267mg,收率为62%。1HNMR(300MHz,CDCl3)δ7.33-7.14(m,6H),7.09-6.95(m,1H),4.57(s,2H),2.41(s,3H),1.99(s,1H).2-溴甲基-4-氟-4’-甲基-1,1’-联苯(4-氟-4’-甲基-[1,1’-联苯基]-2-)甲醇(433mg,2mmol)溶于无水四氢呋喃(10mL)。0℃下,PBr3(146μL,2mmol)逐滴加入反应液中。0℃下继续搅拌2h,加入适量水淬灭反应,然后用乙酸乙酯萃取三次,并用饱和食盐水洗涤,有机相经无水硫酸钠干燥后脱溶,柱层析纯化,得无色液体406mg,收率为73%。1HNMR(300MHz,CDCl3)δ7.32-7.21(m,6H),7.07-7.01(m,1H),4.41(s,2H),2.42(s,3H).(4-氯-[1,1’-联苯基]-2-)甲醇参照(4-氟-4’-甲基-[1,1’-联苯基]-2-)甲醇的方法制备1HNMR(300MHz,CDCl3)δ7.58(d,J=2.1Hz,1H),7.46-7.37(m,3H),7.36-7.29(m,3H),7.20(d,J=8.2Hz,1H),4.59(s,2H).(4’-氯-4-氟-[1,1’-联苯基]甲醇参照(4-氟-4’-甲基-[1,1’-联苯基]-2-)甲醇的方法制备1HNMR(300MHz,CDCl3)δ7.42-7.36(m,2H),7.33-7.16(m,4H),7.03(m,1H),4.57(s,2H).(4-氟-[1,1’-联苯基]-2-)甲醇参照(4-氟-4’-甲基-[1,1’-联苯基]-2-)甲醇的方法制备1HNMR(300MHz,CDCl3)δ7.41(t,J=8.1Hz,3H),7.32(m,3H),7.28-7.22(m,1H),7.05(m,1H),4.61(d,J=5.4Hz,2H).(4’-甲基-[1,’-联苯基]-2-)甲醇参照(4-氟-4’-甲基-[1,1’-联苯基]-2-)甲醇的方法制备1HNMR(300MHz,CDCl3)δ7.57-7.52(m,1H),7.42-7.32(m,2H),7.31-7.21(m,5H),4.63(s,2H),2.42(s,3H).(4-氟-4’-甲氧基-[1,1’-联苯基]-2-)甲醇参照(4-氟-4’-甲基-[1,1’-联苯基]-2-)甲醇的方法制备1HNMR(300MHz,CDCl3)δ7.29-7.18(m,4H),7.04-6.93(m,3H),4.58(d,J=2.8Hz,2H),3.84(s,3H).(4-氟-3’-甲基-[1,1’-联苯基]-2-)甲醇参照(4-氟-4’-甲基-[1,1’-联苯基]-2-)甲醇的方法制备1HNMR(300MHz,CDCl3)δ7.32-7.25(m,2H),7.21-7.17(m,2H),7.10-7.07(m,2H),7.04-6.98(m,1H),4.58(d,J=2.6Hz,2H),2.39(s,3H).[1,1’-联苯基]-2-甲醇参照(4-氟-4’-甲基-[1,1’-联苯基]-2-)甲醇的方法制备1HNMR(300MHz,CDCl3)δ7.57-7.54(m,1H),7.46-7.34(m,7H),7.31-7.26(m,1H),4.62(d,J=1.0Hz,2H).(4,4’-二氯-[1,1’-联苯基]-2-)甲醇参照(4-氟-4’-甲基-[1,1’-联苯基]-2-)甲醇的方法制备1HNMR(300MHz,CDCl3)δ7.57(d,J=1.9Hz,1H),7.40(d,J=8.3Hz,2H),7.32(dd,J=8.2,2.1Hz,1H),7.29-7.22(m,2H),7.17(d,J=8.2Hz,1H),4.56(s,2H).(4-氯-4’-甲基-[1,1’-联苯基]-2-)甲醇参照(4-氟-4’-甲基-[1,1’-联苯基]-2-)甲醇的方法制备1HNMR(300MHz,CDCl3)δ7.54(d,J=2.1Hz,1H),7.22(m,6H),4.56(s,2H),2.39(s,3H).(4-氟-2’-甲基-[1,1’-联苯基]-2-)甲醇参照(4-氟-4’-甲基-[1,1’-联苯基]-2-)甲醇的方法制备3-(3,5-二氟-4-羟基苯基)丙酸乙酯室温下,2,6-二氟苯酚(650mg,5mmol)溶于三氟乙酸(10mL)。向该反应液中分批加入六亚甲基四胺(701mg,5mol)。加毕后,氮气保护下,加热至回流,搅拌过夜。原料消失后,反应液冷去至室温,加入适量二氯甲烷,并依次用水、10%碳酸钾水溶液洗涤,合并水相并用浓盐酸酸化后,乙酸乙酯萃取三次,饱和食盐水洗,无水硫酸钠干燥后脱溶,柱层析纯化,得白色固体。该固体溶于丙酮(10mL)中,并依次加入溴苄(653μL,5.5mmol)、碳酸钾(1.59g,11.5mmol)。加毕后,加热至回流,搅拌过夜。原料消失后,反应液冷却至室温后直接旋干,柱层析纯化,得淡黄色固体390mg,两步收率为31%。在0℃、氮气保护条件下,磷酰基乙酸三乙酯(468μL,2.36mmol)逐滴加入钠氢(88mg,2.2mmol)的甲苯(10mL)溶液。加毕后,升温至50℃。搅拌1h后,反应液冷却至0℃。上一步产物溶于甲苯(5mL)后逐滴加入反应液中。加毕后,加热至回流,搅拌过夜。原料消失后,反应液冷却至室温,加入适量水淬灭反应,乙酸乙酯萃取三次,饱和食盐水洗涤,无水硫酸钠干燥,脱溶、柱层析纯化,得黄色油状物383mg,收率为77%。该产物溶于甲醇(5ml),加入15mg钯碳(含量10%),在氢气氛下反应,搅拌过夜。原料消失后,硅藻土抽滤并用乙酸乙酯洗涤,柱层析纯化,得无色液体228mg,收率为83%。1HNMR(300MHz,CDCl3)δ6.78-6.70(m,2H),5.27(bs,1H),4.12(q,J=7.1Hz,2H),2.84(t,J=7.5Hz,2H),2.56(t,J=7.5Hz,2H),1.23(t,J=7.1Hz,3H).3-(4-羟基-3,5-二甲基苯基)丙酸乙酯室温下,4-羟基-3,5-二甲基苯甲醛(451mg,3mmol)、碳酸钾(829mg,6mmol)、溴苄(428μL,3.6mmol)依次加入N,N-二甲基甲酰胺(10mL)。加毕,反应液在室温搅拌过夜。原料消失后,加入适量水,乙酸乙酯萃取三次,无水硫酸钠干燥,蒸出溶剂,柱层析纯化,得无色液体。在0℃、氮气保护条件下,磷酰基乙酸三乙酯(893μL,4.5mmol)逐滴加入钠氢(168mg,4.2mmol)的甲苯(10mL)溶液。加毕后,升温至50℃。搅拌1h后,反应液冷却至0℃。上一步产物溶于甲苯(5mL)后逐滴加入反应液中。加毕后,加热至回流,搅拌过夜。原料消失后,反应液冷却至室温,加入适量水淬灭反应,乙酸乙酯萃取三次,饱和食盐水洗涤,无水硫酸钠干燥,脱溶、柱层析纯化,得黄色液体。该产物溶于甲醇(15ml),加入90mg钯碳(含量10%),在氢气氛下反应,搅拌过夜。原料消失后,硅藻土抽滤并用乙酸乙酯洗涤,柱层析纯化,得无色液体467mg,三步总收率为70%。1HNMR(300MHz,CDCl3)δ6.80(s,2H),4.56(s,1H),4.12(q,J=7.1Hz,2H),2.81(t,J=7.8 Hz,2H),2.56(t,J=7.8Hz,2H),2.21(s,6H),1.24(t,J=7.1Hz,3H).3-(3,5-二溴-4-羟基苯基)丙酸甲酯室温下,3-(4-羟基苯基)丙酸甲酯(360mg,2.0mmol)溶于冰醋酸(20mL)。向该反应液中逐滴加入液溴(205μL,4.0mmol)。加毕后,室温下搅拌过夜。原料消失后,减压蒸除溶剂,加入饱和亚硫酸钠溶液淬灭反应,乙酸乙酯萃取三次,无水硫酸钠干燥,脱溶,柱层析纯化,得无色液体460mg,收率为68%。1HNMR(300MHz,CDCl3)δ7.28(s,2H),5.79(bs,1H),3.66(s,3H),2.83(t,J=7.5Hz,2H),2.57(t,J=7.6Hz,2H).3-(3,5-二氯-4-羟基苯基)丙酸乙酯室温下,2,6-二氯苯酚(490mg,3mmol)溶于三氟乙酸(10mL)。向该反应液中分批加入六亚甲基四胺(421mg,3mol)。加毕后,氮气保护下,加热至回流,搅拌过夜。原料消失后,反应液冷去至室温,加入适量二氯甲烷,并依次用水、10%碳酸钾水溶液洗涤,合并水相并用浓盐酸酸化后,乙酸乙酯萃取三次,饱和食盐水洗,无水硫酸钠干燥后脱溶,柱层析纯化,得白色固体。该固体溶于N,N-二甲基甲酰胺(5mL)中,并依次加入溴苄(343μL,2.89mmol)、碳酸钾(666mg,4.82mmol)。加毕后,加热至回流,搅拌过夜。原料消失后,反应液冷却至室温后直接旋干,柱层析纯化,得淡黄色固体585mg,两步收率为69%。在0℃、氮气保护条件下,磷酰基乙酸三乙酯(619μL,3.12mmol)逐滴加入钠氢(117mg,2.91mmol)的甲苯(10mL)溶液。加毕后,升温至50℃。搅拌1h后,反应液冷却至0℃。上一步产物溶于甲苯(5mL)后逐滴加入反应液中。加毕后,加热至回流,搅拌过夜。原料消失后,反应液冷却至室温,加入适量水淬灭反应,乙酸乙酯萃取三次,饱和食盐水洗涤,无水硫酸钠干燥,脱溶、柱层析纯化,得白色固体515mg,收率为70%。该产物溶于甲醇(5ml),加入52mg钯碳(含量10%),在氢气氛下反应,搅拌过夜。原料消失后,硅藻土抽滤并用乙酸乙酯洗涤,柱层析纯化,得无色液体254mg,收率为66%。1HNMR(300MHz,CDCl3)δ7.11(s,2H),5.78(s,1H),4.13(q,J=7.2Hz,2H),2.84(t,J=7.5Hz,2H),2.57(t,J=7.5Hz,2H),1.24(t,J=7.1Hz,3H).2,6-二氟-4-(2-羟基乙氧基)苯酚0℃条件下,NBS(890mg,5mmol)分批加入2,6-氟苯酚(650mg,5mmol)的DMF溶液中。加毕后,升至室温搅拌过夜。原料消失后,反应液直接柱层析纯化,得黄色液体,直接用于下 一步反应。上一步所得液体溶于DMF中,依次加入溴苄(713μL,6mmol)、碳酸钾(1.38mg,10mmol)。加毕后反应液在室温下搅拌过夜。原料消失后,往反应液中加入适量水,乙酸乙酯萃取三次后无水硫酸钠干燥,减压蒸除溶剂后柱层析纯化,得无色液体1.28g,两步收率85%。在斯莱克管中依次加入上述产物(300mg,1mmol)、氯化铜的水合物(8.5mg,0.05mmol)、碳酸钾(410mg,3mmol)和乙二醇(5mL)。加毕后,反应液在氩气保护下,升温至130℃,搅拌过夜。原料消失后,加入3MHCl溶液酸化至酸性,乙酸乙酯萃取,减压蒸出溶剂后,柱层析纯化,得无色液体175mg,收率63%。上一步所得产物(411mg,1.47mmol)溶于甲醇(5mL)中,加入10%的钯碳(40mg),在氢气氛下,室温搅拌过夜。原料消失后,硅藻土滤除钯碳,滤液减压蒸除溶剂后,柱层析纯化,得白色固体238mg,收率85%。1HNMR(300MHz,CDCl3)δ6.53(d,J=8.2Hz,2H),3.98(m,4H).实施例13-(3,5-二氟-4-((4-氟-4’-甲基-[1,1’-联苯基]-2-)甲氧基)苯基)丙酸(I-1)室温下,(4-氟-4’-甲基-[1,1’-联苯基]-2-)甲醇(160mg,0.74mmol)、3-(3,5-二氟-4-羟基苯基)丙酸乙酯(170mg,0.74mmol)、三丁基膦(295μL,1.18mmol)溶于无水甲苯(10mL)。N2保护下,向上述反应液中逐滴加入ADDP(298mg,1.18mmol)的甲苯(5mL)溶液。加毕后,反应液在室温下搅拌过夜。原料消失后,向反应瓶中加入适量正己烷,搅拌10min后硅藻土抽滤并用正己烷洗涤滤饼。减压蒸除溶剂后,柱层析纯化,得无色液体。该无色液体溶于甲醇(2mL),饱和氢氧化钠溶液(1mL)加入混合物中,室温下搅拌过夜。原料消失后,减压蒸出部分溶剂后用3M稀盐酸酸化至酸性,乙酸乙酯萃取三次,无水硫酸钠干燥,脱溶,柱层析纯化,得白色固体80mg,两步收率为27%。1HNMR(300MHz,CDCl3)δ7.40(dt,J=12.1,6.0Hz,1H),7.33-7.18(m,5H),7.08(td,J=8.4,2.7Hz,1H),6.84-6.67(m,2H),5.00(s,2H),2.89(t,J=7.5Hz,2H),2.66(t,J=7.5Hz,2H),2.41(s,3H).13CNMR(75MHz,CDCl3)δ178.05,163.70,160.44,157.66,157.57,154.36,154.28,137.75,137.71,137.12,136.44,136.30,136.19,136.15,136.05,131.60,131.49,129.17,128.95,116.10,115.81,115.54,115.23,114.95,112.12,112.02,111.91,111.81,73.42,34.88,29.76,21.12.实施例23-(4-((4-氯-[1,1’-联苯基]-2-)甲氧基)-3,5-二氟苯基)丙酸(I-2)参照实施例1的方法制备。1HNMR(300MHz,CDCl3)δ7.68(d,J=1.0Hz,1H),7.44-7.35(m,6H),7.26(d,J=4.1Hz,1H),6.72(m,2H),4.97(s,2H),2.87(t,J=7.4Hz,2H),2.65(t,J=7.4Hz,2H).13CNMR(75MHz,CDCl3)δ178.56,157.62,157.54,155.19,154.32,154.24,140.40,139.11,136.39,136.28,136.17,135.55,135.18,133.38,131.26,130.18,130.01,129.86,129.62,129.33,129.15,128.39,128.26,128.08,127.56,127.48,127.20,112.10,112.00,111.89,111.80,111.72,73.22,34.91,29.70.实施例33-(4-((4’-氯-4-氟-[1,1’-联苯基]-2-)甲氧基)-3,5-二氟苯基)丙酸(I-3)参照实施例1的方法制备。实施例43-(3,5-二氟-4-((4-氟-[1,1’-联苯基]-2-)甲氧基)苯基)丙酸(I-4)参照实施例1的方法制备。13CNMR(75MHz,CDCl3)δ178.50,163.81,160.55,157.66,157.58,154.36,154.28,139.38,137.88,137.84,136.43,136.32,136.20,136.16,136.06,131.61,131.51,129.33,128.27,127.43,116.27,115.98,115.33,115.05,112.15,112.05,111.94,111.85,73.36,34.95,29.76.实施例53-(3,5-二氟-4-((4’-甲基-[1,1’-联苯基]-2-)甲氧基)苯基)丙酸(I-5)参照实施例1的方法制备。1HNMR(300MHz,CDCl3)δ7.72-7.59(m,1H),7.46-7.32(m,5H),7.25(d,J=7.7Hz,2H),6.75(d,J=8.6Hz,2H),5.04(s,2H),2.89(t,J=7.4Hz,2H),2.67(t,J=7.4Hz,2H),2.43(s,2H).13CNMR(75MHz,CDCl3)δ178.03,157.81,157.73,154.51,154.43,154.35,154.33,154.28,154.25,142.28,137.41,136.92,136.88,136.03,135.93,133.73,130.09,129.25,128.83,128.41,128.37,128.24,127.31,112.05,111.95,111.84,111.74,74.02,34.91,29.77,21.15.实施例63-(3,5-二氟-4-((4-氟-4’-甲氧基-[1,1’-联苯基]-2-)甲氧基)苯基)丙酸(I-6)参照实施例1的方法制备。1HNMR(300MHz,CDCl3)δ7.37(dd,J=9.6,2.5Hz,1H),7.33-7.21(m,3H),7.07(td,J=8.3,2.6Hz,1H),6.94(d,J=8.6Hz,2H),6.79-6.68(m,2H),4.98(s,2H),3.85(s,3H),2.88(t,J=7.5Hz,2H),2.65(t,J=7.5Hz,2H).13CNMR(75MHz,CDCl3)δ177.85,163.61,160.35,159.03,157.66,157.58,154.36,154.28,137.63,137.56,137.52,136.33,136.22,136.14,136.04,131.77,131.65,131.55,131.41,130.43,116.23,115.94,115.28,115.00,114.59,113.70,112.13,112.03,111.92,111.83,73.48,55.29,34.85,29.77.实施例73-(3,5-二氟-4-((4-氟-3’-甲基-[1,1’-联苯]-2-)甲氧基)苯基)丙酸(I-7)参照实施例1的方法制备。1HNMR(300MHz,CDCl3)δ7.42(dd,J=9.5,2.1Hz,1H),7.29(dd,J=14.8,6.8Hz,2H),7.22-7.05(m,4H),6.75(d,J=8.6Hz,2H),4.99(s,2H),2.88(t,J=7.4Hz,2H),2.66(t,J=7.4Hz,2H),2.40(s,3H).13CNMR(75MHz,CDCl3)δ178.41,170.78,163.74,160.48,157.66,157.57,154.36,154.28,139.32,138.98,138.00,137.96,137.91,136.32,136.21,136.10,136.00,131.55,131.45,130.04,128.14,128.12,126.37,116.24,115.95,115.26,114.98,112.12,112.02,111.92,111.82,73.40,34.93,29.75,21.35.实施例83-(4-([1,1’-联苯]-2-甲氧基)-3,5-二氟苯基)丙酸(I-8)参照实施例1的方法制备。1HNMR(300MHz,CDCl3)δ7.70-7.57(m,1H),7.48-7.30(m,8H),6.79-6.66(m,2H),5.00(s,2H),2.86(t,J=7.5Hz,2H),2.64(t,J=7.5Hz,2H).实施例93-(4-((4,4’-二氯-[1,1’-联苯基]-2-)甲氧基)-3,5-二氟苯基)-丙酸(I-9)参照实施例1的方法制备。1HNMR(300MHz,CDCl3)δ7.62(d,J=1.7Hz,1H),7.43-7.30(m,4H),7.27-7.19(m,2H),6.74(d,J=8.8Hz,2H),4.90(s,2H),2.87(t,J=7.5Hz,2H),2.64(t,J=7.5Hz,2H).实施例103-(4-氯-4’-甲基-[1,1’-联苯基]-2-)-甲氧基)-3,5-二氟苯基)丙酸(I-10)参照实施例1的方法制备。1HNMR(300MHz,CDCl3)δ7.67(s,1H),7.42-7.18(m,6H),6.75(d,J=8.7Hz,2H),4.98(s,2H),2.89(t,J=7.3Hz,2H),2.66(t,J=7.3Hz,2H),2.41(s,3H).实施例113-(3,5-二氟-4-((4-氟-2’-甲基-[1,1’-联苯基]-2-)甲氧基)苯基)丙酸(I-11)参照实施例1的方法制备。1HNMR(300MHz,CDCl3)δ7.48(d,J=9.7Hz,1H),7.27-7.04(m,6H),6.71(d,J=8.7Hz,2H),4.80(s,2H),2.87(t,J=7.4Hz,2H),2.64(t,J=7.4Hz,2H),2.06(s,3H).13CNMR(75MHz,CDCl3)δ178.02,163.82,160.57,157.56,157.48,154.26,154.18,138.69,136.97,136.87,136.41,136.36,136.24,136.13,136.10,136.02,131.10,131.00,129.97,129.71,127.79,125.57,115.24,114.92,114.64,114.58,112.08,111.99,111.88,111.78,73.01,34.88,29.74,19.85.实施例123-(4-((4-氟-4’-甲基-[1,1’-联苯基]-2-)甲氧基)-3,5-二甲基苯基)丙酸(I-12)参照实施例1的方法制备。1HNMR(300MHz,CDCl3)δ7.52-7.49(m,1H),7.23-7.20(m,1H),7.17(bs,4H),7.08-7.02(m,1H),6.81(s,2H),4.68(s,2H),2.83(t,J=7.7Hz,2H),2.62(t,J=7.7Hz,2H),2.37(s,3H),2.12(s,6H).13CNMR(75MHz,CDCl3)δ178.93,163.93,160.67,154.11,137.75,137.65,137.08,136.97,136.93,136.76,135.70,131.48,131.37,131.04,129.07,129.00,128.62,115.04,114.74,114.48,114.19,70.97,35.68,29.92,21.12,16.31.实施例133-(3,5-二溴-4-((4-氟-4’-甲基-[1,1’-联苯基]-2-)甲氧基)苯基)丙酸(I-13)室温下,2-溴甲基-4-氟-4’-甲基-1,1’-联苯(74mg,0.27mmol)、3-(3,5-二溴-4-羟基苯基)丙酸甲酯(90mg,0.27mmol)溶于DMF中。向上述反应液中加入碳酸钾(74mg,0.53mmol)。加毕后,室温搅拌过夜。原料消失后,加入适量水,乙酸乙酯萃取三次后无水硫酸钠干燥,脱溶,柱层析纯化,得无色油状物。该无色液体溶于甲醇(2mL),饱和氢氧化钠溶液(1mL)加入混合物中,室温下搅拌过夜。原料消失后,减压蒸出部分溶剂后用3M稀盐酸酸化至酸性,乙酸乙酯萃取三次,无水硫酸钠干燥,脱溶,柱层析纯化,得白色固体124mg,两步收率为88%。实施例143-(3,5-二氯-4-((4-氟-4’-甲基-[1,1’-联苯]-2-)甲氧基)苯基)丙酸(I-14)参照实施例13的方法制备。1HNMR(300MHz,CDCl3)δ7.59(dd,J=9.8,2.6Hz,1H), 7.31-7.19(m,5H),7.17-7.05(m,3H),4.93(s,2H),2.88(t,J=7.6Hz,2H),2.67(t,J=7.5Hz,2H),2.41(s,3H).实施例152-(3,5-二氟-4-((4-氟-4’-甲基-[1,1’-联苯基]-2-)甲氧基)苯氧基)乙酸(I-15)室温下,2-溴甲基-4-氟-4’-甲基-1,1’-联苯(140mg,0.5mmol)、2,6-二氟-4-(2-羟基乙氧基)苯酚(95mg,0.5mmol)、碳酸钾(104mg,0.75mmol)溶于DMF中,搅拌过夜。原料消失后,加入适量水,乙酸乙酯萃取三次,减压蒸除溶剂,柱层析纯化,得无色液体87mg。上述产物溶于丙酮(5mL)中,冰浴条件下,逐滴加入Jones试剂(1mL),继续搅拌过夜。原料消失后,加入适量异丙醇至体系呈蓝色,乙酸乙酯萃取三次,减压蒸除溶剂,直接柱层析纯化,得白色固体53mg,两步收率为26%。1HNMR(300MHz,CDCl3)δ7.39(d,J=9.4Hz,1H),7.32-7.18(m,5H),7.08(t,J=8.4Hz,1H),6.49(d,J=9.0Hz,2H),4.93(s,2H),4.61(s,2H),2.41(s,3H).实施例162-(3,5-二氟-4-((4’-甲基-[1,1’-联苯基]-2-)甲氧基)苯氧基)乙酸(I-17)参照实施例15的方法制备。1HNMR(300MHz,CDCl3)δ7.68-7.58(m,1H),7.38(ddd,J=12.1,9.2,5.2Hz,5H),7.24(d,J=7.8Hz,2H),6.56-6.41(m,2H),4.98(s,2H),4.62(s,2H),2.42(s,3H).体外GPR120激动活性药效学试验药品:受试化合物(上述实施例制备)、对照药TUG-891(3-(4-((4-氟-4’-甲基-[1,1’-联苯基]-2-)甲氧基)苯基)丙酸)、GW9508(3-(4-((3-苯氧基苄基)氨基)苯基)丙酸)、α-LA。细胞模型:利用细胞转基因技术,在CHO-K1细胞中过表达hGPR120与Gα15蛋白,经过筛选、扩大培养、鉴定,得到人工构建的hGPR120稳定过表达细胞系。进一步利用受体稳定过表达的细胞系,通过检测细胞内Ca2+浓度变化,在细胞水平对小分子化合物的GPR120的激动活性分别进行评价。实验步骤:hGPR120稳定过表达细胞系的构建委托Invitrogen公司构建hGPR120质粒,载体均为pcDNA3.1。使用罗氏X-tremGENEHP转染试剂对CHO-K1细胞进行转染。采用抗生素筛选结合有限稀释法,得到稳定过表达 hGPR120的单克隆细胞株。RT-PCR鉴定单克隆细胞株中受体的表达情况,验证蛋白的表达。检测内源性配体α-LA在构建的hGPR120稳定过表达细胞系的激动活性,验证细胞的功能性表达。化合物准备化合物用DMSO配成100mM的母液储存钙流实验步骤1)细胞铺板:细胞以3×104/孔的密度接种至96孔板,置于37℃、5%CO2的细胞培养箱过夜培养;2)弃去培养基,每孔加入100ulHBSS清洗后,加入100uL含Probenecid的Fluo-4染料溶液37℃孵育60min。3)孵育结束后,吸出Fluo-4染料溶液,加100μLHBSS缓冲液,洗去染料;4)每孔加入100μL含Probenecid的HBSS,37℃孵育10min,;5)准备化合物板:96孔板中每孔加入至少150μL浓度为3×工作浓度的化合物,HBSS配制;6)加入相应浓度的化合物溶液;7)读数:按照参数设置表用FlexStation3读数。FlexStation参数设置表(96孔板)参数Excitationwavelentth(nm)485Emissionwavelength(nm)525Emissioncut-off(nm)515Dyeloadingvolume(μL)100Additionvolume(μL)50Pipetheight(μL)100实施例17片剂将实施例1中制得的化合物I-1(50g)、羟丙甲基纤维素E(150g)、淀粉(200g)、聚维酮K30适量和硬脂酸镁(1g)混合,制粒,压片。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1