过渡金属配合物的制备方法与流程
本发明涉及烯烃共聚物制备用过渡金属配合物的制备方法,更具体地,涉及包含元素周期表上的第四族的过渡金属、环戊二烯配体及可通过升华或单纯过滤来实现提纯的至少一个酚类配体的过渡金属配合物的制备方法,本发明的特征在于,在制备过渡金属配合物的过程中,起始原料不包含卤素,尤其不包含氯。
背景技术:
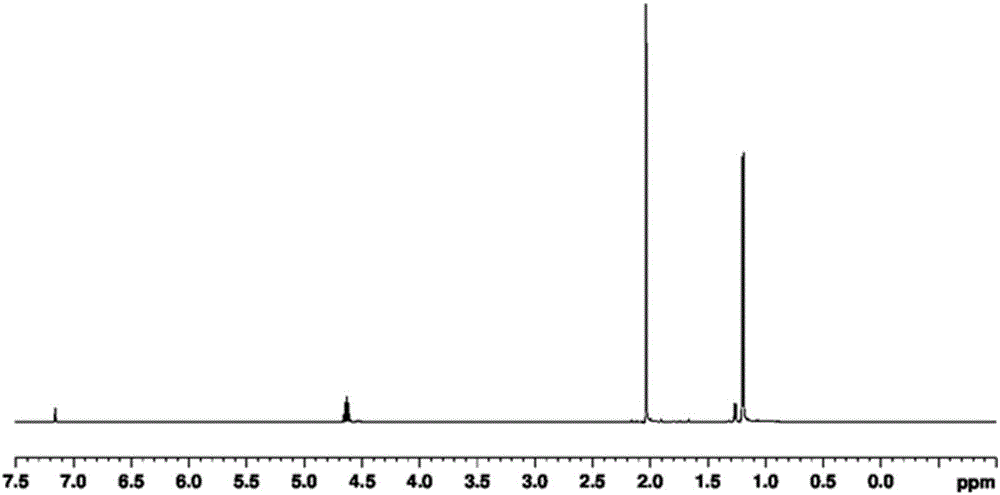
:元素周期表上的第四(IV)族的过渡金属配合物作为用于制备聚乙烯的过渡金属催化剂而被广泛使用。尤其,包含环戊二烯的过渡金属配合物一直适用于制备各种高分子,并且利用酚类配体并通过文献中二已知的方法来合成。为了定量注入具有高分子制备工序中的活性的催化剂以及控制具有非活性的变性种,在制备催化剂的过程中,异物的管理及查明是非常重要的部分。尤其,由于对水分敏感的元素周期表上的第四(IV)族的过渡金属的特征,正非常严格地实施借助水分的变性种的管理。并且,为了定量注入过渡金属催化剂,在制备过渡金属催化剂过程中,去除残留配体是非常重要的部分。在过渡金属配合物留下残留苯酚有机物的余量时,则会生成作为杂质的催化剂变形物质以及很难实现精确的催化剂注入量,因而存在很难调节精密高分子反应的问题。在元素周期表上的第四(IV)族的过渡金属配合物和酚类配体的情况下,通过再结晶的分离为普通的方法,但可能会带来制备成本的增加,因此,目前需要更有效的提纯方法。并且,若使用可腐蚀用于制备聚乙烯的工序上的材质的氯化合物,则对上述氯化合物实施严格的管理,由此特别需要注意催化剂内的氯化合物的含量管理。因此,目前急需开发在制备催化剂的过程中使借助水分的变性种最小化、可有效地去除残留配体、起始原料不包含氯的过渡金属配合物的制备方法。现有技术文献非专利文献(非专利文献1)Inorg.Chem.1989,28(10),pp2003-2007(非专利文献2)J.Organomet.Chem.1997,544,pp207-215技术实现要素:技术问题为了克服上述现有技术的问题而进行研究的结果,本发明人发现,作为起始原料,若使不包含卤素的过渡金属醇盐前体,尤其使不包含氯的过渡金属醇盐前体和可通过升华或单纯过滤来实现提纯的酚类配体发生反应,则不仅可使借助水分的变性种的制备最小化,还可制备不包含氯的过渡金属配合物,由此完成了本发明。因此,本发明的目的在于,提供如下的过渡金属配合物的制备方法,即,使不包含卤素的过渡金属醇盐前体,尤其使不包含氯的过渡金属醇盐前体和可通过升华或单纯过滤来实现提纯的酚类配体发生反应来制备过渡金属配合物。解决问题的方案本发明涉及烯烃共聚物制备用过渡金属配合物的制备方法,更具体地,涉及如下的过渡金属配合物的制备方法,即,使下述化学式2的过渡金属醇盐前体和下述化学式3的酚类配体发生反应来制备下述化学式1的过渡金属配合物,上述化学式1的过渡金属配合物包含元素周期表上的第四族的过渡金属、环戊二烯配体、可通过升华或单纯过滤来实现提纯的至少一个酚类配体,本发明的特征在于,在制备过渡金属配合物的过程中,起始原料不包含卤素,尤其不包含氯。化学式1:化学式2:化学式3:在上述化学式1、2及3中,M为元素周期表上的第四族的过渡金属;Cp为能够与M形成η5-键环戊二烯环或包含环戊二烯环的稠环,上述环戊二烯环或包含环戊二烯环的稠环还能够被选自(C1–C20)烷基、(C6–C30)芳基、(C2–C20)烯基及(C6–C30)芳基、(C1–C20)烷基中的一种以上取代;R1为(C1–C20)烷基;R2、R3、R4、R5及R6分别独立地为氢、卤素、(C1–C30)烷基、(C6–C30)芳基、(C6–C30)芳基(C1–C30)烷基、(C1–C30)烷基(C6–C30)芳基、(C1–C30)烷氧基(C6–C30)芳氧基或NR'R”,R2和R3或R5和R6分别与(C2–C6)亚烃基或(C2–C6)亚烯基相连接来形成稠环;R'和R”分别独立地为(C1–C30)烷基或(C6–C30)芳基;n为1至3的整数。在本发明一实施例的过渡金属配合物的制备方法中,上述反应可在有机溶剂中或纯净(neat)的条件下实现,只要可溶解上述反应物质,则对有机溶剂不进行限制。在本发明一实施例的过渡金属配合物的制备方法中,上述反应可在上述溶剂的回流温度范围内进行。在本发明一实施例的过渡金属配合物的制备方法中,上述化学式2的过渡金属醇盐前体与上述化学式3的酚类配体的摩尔比可以是1:1.1至1:3.5。在本发明一实施例的过渡金属配合物的制备方法中,上述R2、R3、R5及R6可分别独立地选自氢、卤素、(C1–C30)烷基、(C6–C30)芳基或者R2和R3或R5和R6可分别与(C2–C6)亚烃基或(C2–C6)亚烯基相连接来形成稠环;R4可以为氢、卤素、(C1–C30)烷基、(C1–C30)烷氧基、(C6–C30)芳氧基或NR'R”;R'及R”可分别独立地为(C1–C30)烷基或(C6–C30)芳基。在本发明一实施例的过渡金属配合物的制备方法中,可使下述化学式2的过渡金属醇盐前体和下述化学式3的酚类配体发生反应来制备下述化学式4的过渡金属配合物。化学式4:化学式2:化学式3:在上述化学式2、3及4中,M为元素周期表上的第四族的过渡金属;Cp为可M形成η5-键的环戊二烯环或环戊二烯环的稠环,上述环戊二烯环或包含环戊二烯环的稠环还可以被选自(C1–C20)烷基、(C6–C30)芳基、(C2–C20)烯基及(C6–C30)芳基(C1–C20)烷基中的一种以上取代;R1为(C1–C20)烷基;R2及R6分别独立地为氢、卤素、(C1–C20)烷基或(C6–C20)芳基;R3及R5分别独立地为氢或卤素;R2和R3或R5和R6可分别与(C2–C6)亚烃基或(C2–C6)亚烯基相连接来形成稠环;R4为氢、卤素、(C1–C20)烷基、(C1–C20)烷氧基或二(C1–C20)烷基氨基。在本发明一实施例的过渡金属配合物的制备方法中,上述化学式2的过渡金属醇盐前体与上述化学式3的酚类配体的摩尔比可以是1:3.0至1:3.5。在本发明一实施例的过渡金属配合物的制备方法中,上述化学式4的过渡金属配合物可以为选自下述结构中的过渡金属配合物:在本发明一实施例的过渡金属配合物的制备方法中,在使上述化学式2的过渡金属醇盐前体和上述化学式3的酚类配体发生反应之后,为了去除未反应的酚类配体,本发明的过渡金属配合物的制备方法还可包括通过升华或单纯过滤来实现提纯的过程。发明的效果在本发明的过渡金属配合物的制备方法中,作为起始原料,可通过使不包含卤素过渡金属醇盐前体,尤其不包含氯的过渡金属醇盐前体和可通过升华或单纯过滤来实现提纯的酚类配体发生反应来以高收率制备过渡金属配合物,由此本发明具有如下的优点,即,不仅可使作为现有问题的借助水分的变性种的形成最小化,还可通过升华或单纯过滤提纯简单地分离所制备的过渡金属配合物和未反应的酚类配体。并且,由于在制备过渡金属配合物过程中不包含卤素,尤其完全不包含氯,因此即使将所制备的过渡金属配合物利用在烯烃聚合,也不用担心腐蚀工序上的材质,并且在作为以往使用的起始原料的过渡金属氯化物前体的情况下,由于必须需要同时使用胺类化合物,因此使生成物和胺残物共存,但在作为起始原料使用过渡金属醇盐前体而不是氯化物的本发明的情况下,具有不使胺残物等的杂质共存的优点。并且,由于反应选择性优秀,具有可以仅通过反应物质的摩尔比变化来简单地制备结合有1、2或3个酚类配体的过渡金属配合物的优点。附图说明图1为Cp*Ti(OMe)3的1H-NMR数据。图2为Cp*Ti(iOPr)3的1H-NMR数据。图3为Cp*Ti(OMe)3和3.1当量的4–t–辛基苯酚(4-tert-Octylphenol)的反应中的升华前的1H-NMR数据。图4为Cp*Ti(OMe)3和3.1当量的4–t–辛基苯酚的反应中的升华后的1H-NMR数据。图5为Cp*Ti(iOPr)3和3.1当量的4–t–辛基苯酚的反应中的升华后的1H-NMR数据。图6为比较例1的Cp*TiCl和3.1当量的4–t–辛基苯酚的反应后的1H-NMR数据。图7为比较例2的Cp*TiCl、3.2当量的三乙胺和3.1当量的4–t–辛基苯酚的反应后的1H-NMR数据。具体实施方式本发明涉及烯烃共聚物制备用过渡金属配合物的制备方法,更具体地,提供如下的过渡金属配合物的制备方法,即,使下述化学式2的过渡金属醇盐前体和下述化学式3的酚类配体发生反应来制备下述化学式1的过渡金属配合物,上述化学式1的过渡金属配合物包含元素周期表上的第四族的过渡金属、环戊二烯配体、可通过升华或单纯过滤来实现提纯的至少一个酚类配体。本发明的过渡金属配合物的制备方法的特征在于,在制备乙烯单独聚合物或乙烯和α–烯烃的共聚物制备用渡金属配合物的过程中,不包含作为杂质的卤素,尤其不包含氯。化学式1:化学式2:化学式3:在上述化学式1、2及3中,M为元素周期表上的第四族的过渡金属;Cp为能够与M形成η5-键的环戊二烯环或包含环戊二烯环的稠环,上述环戊二烯环或包含环戊二烯环的稠环还能够被选自(C1–C20)烷基、(C6–C30)芳基、(C2–C20)烯基及(C6–C30)芳基、(C1–C20)烷基中的一种以上取代;R1为(C1–C20)烷基;R2、R3、R4、R5及R6分别独立地为氢、卤素、(C1–C30)烷基、(C6–C30)芳基、(C6–C30)芳基(C1–C30)烷基、(C1–C30)烷基(C6–C30)芳基、(C1–C30)烷氧基(C6–C30)芳氧基或NR'R”,R2和R3或R5和R6分别与(C2–C6)亚烃基或(C2–C6)亚烯基相连接来形成稠环;R'和R”分别独立地为(C1–C30)烷基或(C6–C30)芳基;n为1至3的整数。上述“烷基”均包含直链上或支链上的碳链。在本发明中所制备的化学式1的过渡金属配合物作为在第四族过渡金属周围包含环戊二烯基配体至少一个芳氧基配体芳氧基配体且配体间相互未交联的结构,在制备乙烯单独聚合物或乙烯和α–烯的共聚物的过程中,在高温度的条件下也具有高活性。在上述化学式1中,若过渡金属为元素周期表上的第四族的过渡金属,则全都可以使用,但优选地,可以为钛、锆或铪,更优选地,可以为钛。在本发明一实施例的过渡金属配合物的制备方法中,上述反应可在有机溶剂中或纯净的条件下实现,只要可溶解上述反应物质,则对有机溶剂不进行限制。纯净的条件是指不使用有机溶剂,而是通过混合上述化学式2的过渡金属醇盐前体和上述化学式3的酚类配体来进行上述反应,因此可在真空条件下进行。考虑到最终化合物的溶解度,优选地,上述有机溶剂使用选自由甲基环已烷(MCH)、己烷、二氯甲烷、甲苯、环己烷、苯及庚烷组成的组中的单独或两个以上的混合溶剂,更优选地使用甲基环已烷或甲苯。根据本发明一实施例的过渡金属配合物的制备方法,优选地,上述反应执行在上述有机溶剂的回流温度。若上述反应执行在未满或超过有机溶剂的回流温度,则可具有不易发生所目的地反应,导致化学式1的过渡金属配合物的收率下降或发生其他副反应的问题。在本发明一实施例的过渡金属配合物的制备方法中,相对于上述化学式2的过渡金属醇盐前体,化学式3的酚类配体的使用量可相同或过量,但在为了防止生成配体数量不同的各种混合物的方面,优选地,上述化学式2的过渡金属醇盐前体与上述化学式3的酚类配体的摩尔比为1:1.1至1:3.5。在本发明一实施例的过渡金属配合物的制备方法中,优选地,上述R2、R3、R5及R6可分别独立地为氢、卤素、(C1–C30)烷基、(C6–C30)芳基或者R2和R3或R5和R6可分别与(C2–C6)亚烃基或(C2–C6)亚烯基相连接来形成稠环;R4为氢、卤素、(C1–C30)烷基、(C1–C30)烷氧基、(C6–C30)芳氧基或NR'R”;R'及R”分别独立地为(C1–C30)烷基或(C6–C30)芳基。在本发明一实施例的过渡金属配合物的制备方法中,由于上述化学式1中的n为3的情况下的具有3个芳氧基配体的过渡金属配合物的空间位阻大,因此在高温条件下活性非常高,从而能够以高收率制备高分子量、低密度的聚合物,由此最适合适用于活性非常高的烯烃共聚物制备用催化剂。更优选地,由于具有优秀的催化活性,因此在制备高分子量及低密度的烯烃共聚物的方面,使下述化学式2的过渡金属醇盐前体和下述化学式3的酚类配体发生反应来制备下述化学式4的过渡金属配合物。化学式4:化学式2:化学式3:在上述化学式2、3及4中,M为元素周期表上的第四族的过渡金属;Cp为可M形成η5-键的环戊二烯环或环戊二烯环的稠环,上述环戊二烯环或包含环戊二烯环的稠环还可以被选自(C1–C20)烷基、(C6–C30)芳基、(C2–C20)烯基及(C6–C30)芳基(C1–C20)烷基中的一种以上取代;R1为(C1–C20)烷基;R2及R6分别独立地为氢、卤素、(C1–C20)烷基或(C6–C20)芳基;R3及R5分别独立地为氢或卤素;R2和R3或R5和R6可分别与(C2–C6)亚烃基或(C2–C6)亚烯基相连接来形成稠环;R4为氢、卤素、(C1–C20)烷基、(C1–C20)烷氧基或二(C1–C20)烷基氨基。用于制备上述化学式4的过渡金属配合物的上述化学式2的过渡金属醇盐前体与上述化学式3的酚类配体的摩尔比为1:3.0至1:3.5,优选地,可以为1:3.0至1:3.1。优选地,上述化学式4的过渡金属配合物可选自如下结构中的过渡金属配合物:在本发明一实施例的过渡金属配合物的制备方法中,为了提高所制备的化学式1的过渡金属配合物的纯度,还可包括如下的步骤,即,在使上述化学式2的过渡金属醇盐前体和上述化学式3的酚类配体发生反应后,从作为生成物的化学式1的过渡金属配合物去除化学式2的未反应的残留酚类配体。去除上述化学式2的未反应的残留酚类配体的步骤如下,即,对在提纯温度为100℃至130℃且0.1至2.0torr的低压条件下未发生反应而残留的酚类配体进行升华或者通过单纯过滤来进行去除,优选地,通过升华来去除未反应的残留酚类配体。可将通过本发明的制备方法来制备的过渡金属配合物作为催化剂使用于烯烃聚合,烯烃聚合方法可利用已公知的方法。参照下述实施例对本发明的效果进行具体的说明。但是,下述实施例仅用于例示本发明,并不用于限制本发明的范围。除非另有说明,本发明的所有催化剂合成实验在氮气氛下利用标准希莱克(Schlenk)技术或手套箱技术来进行,反应中所使用的有机溶剂在金属钠和苯酮条件下以回流的方式除去水分后,在使用前通过蒸馏后使用。所合成的催化剂的1H-NMR分析在常温中使用Bruker500MHz来进行。在使作为聚合溶剂的甲基环己烷经过填充有分子筛和活性氧化铝的管后,利用高纯度的氮以起泡的方式充分地除去水分、氧及其他催化毒物质后使用上述甲基环己烷。完成聚合的聚合物根据以下说明的方法进行分析。[实施例1]利用Cp*Ti(OMe)3的Cp*Ti(4–t–辛基苯酚(4-t-Octylphenolate))3的制备在反应容器混合三甲氧基(五甲基环戊二烯)钛(IV)(Cp*Ti(OMe)3,0.552g,1当量)和4–t–辛基苯酚(1.238g,3.1当量)后,投入甲苯(50mL)。向反应容器连接冷凝器后,回流搅拌12小时。若黄色的反应溶液变成橙色,则在0.5torr的条件下慢慢地去除作为反应溶剂的甲苯后,加热至110度来去除未反应的残留4–t–辛基苯酚,然后,将反应机的温度降低至室温,并定量获得橙色固体的Cp*Ti(4–t–辛基苯酚)3(1.62g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R2=R3=R4=R6=氢,R4=t-辛基),这是由1H-NMR确认的。图1示出了Cp*Ti(OMe)3的1H-NMR数据,图3及图4分别示出了Cp*Ti(OMe)3和3.1当量的4–t–辛基苯酚的反应中的升华前及升华后的生成物Cp*Ti(4–t–辛基苯酚)3的1H-NMR数据,由此得知,比起升华前,升华后催化剂的纯度增加。[实施例2]利用Cp*Ti(OiPr)3的Cp*Ti(4–t–辛基苯酚)3的制备除了代替三甲氧基(五甲基环戊二烯)钛(IV)(Cp*Ti(OMe)3而使用三异丙氧基(五甲基环戊二烯)钛(IV)(Cp*Ti(OiPr)3)以外,以与上述实施例1相同的方法发生反应并定量获得Cp*Ti(4–t–辛基苯酚)3(1.62g)。图2示出Cp*Ti(iOPr)3的1H-NMR数据,图5分别示出使Cp*Ti(iOPr)3和3.1当量的4–t–辛基苯酚发生反应并实现升华后的生成物Cp*Ti(4–t–辛基苯酚)3的1H-NMR数据。[实施例3]利用Cp*Ti(OMe)3的Cp*Ti(苯酚(Phenolate))3的制备在反应容器混合三甲氧基(五甲基环戊二烯)钛(IV)((Cp*Ti(OMe)3,0.552g,1当量)和苯酚(0.583g,3.1当量)后,投入甲基环己烷(Methylcyclohexane,50mL)。向反应容器连接冷凝器(condenser)后,回流搅拌12小时。确认到黄色的反应溶液变成橙色并生成了固体。通过过滤定量获得橙色固体的Cp*Ti(苯酚)3(0.95g)(在化学式1中,M=T,Cp=五甲基环戊二烯,n=3,R2=R3=R4=R5=R6=氢),这是通过1H-NMR和13C-NMR确认的。1H-NMRinCDCl3-d1:δ=7.28(2H,t),,7.01(1H,,d),,6.95(2H,d),2.18(15H,,s);;13C-NMRinCDCl3-d1::δ=157.3,130.5,,130.1,,121.3,,115.9,9.7[实施例4]利用Cp*Ti(OiPr)3的Cp*Ti(苯酚)3的制备除了代替三甲氧基(五甲基环戊二烯)钛(IV)(Cp*Ti(OMe)3而使用三异丙氧基(五甲基环戊二烯)钛(IV)(Cp*Ti(OiPr)3)以外,以与上述实施例3相同的方法发生反应并定量获得Cp*Ti(苯酚)3(0.95g)。[实施例5]利用Cp*Ti(OMe)3的Cp*Ti(2,6-二甲基苯酚(2,6-dimethylphenolate))3的制备除了代替苯酚使用2,6-二甲基苯酚(0.79g,6.3毫摩尔)以外,以与上述实施例3相同的方法发生反应并定量获得橙色固体的Cp*Ti(2,6-二甲基苯酚)3(1.12g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R3=R5=R4=氢,R2=R6=甲基),这是通过1H-NMR和13C-NMR来确认的。1H-NMRinCDCl3-d1:δ=6.99(1H,,m),,6.87(2H,,d),,2.18(15H,,s),2.15(18H,,s);;13C-NMRinCDCl3-d1::δ=158.7,,130.5,,129.0,,126.0,124.3,,15.4,,9.7[实施例6]利用Cp*Ti(OMe)3的Cp*Ti(2,6-二异丙基苯酚(2,6-diisopropylphenolate))3的制备除了代替苯酚而使用2,6-二异丙基苯酚(1.2g,6.3毫摩尔)之外,以与上述实施例3相同的方法发生反应并定量获得橙色固体的Cp*Ti(2,6-二异丙基苯酚)3(1.43g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R3=R5=R4=氢,R2=R6=异丙基),这是通过1H-NMR来确认的。1H-NMRinCDCl3-d1::δ=7.17(2H,d),,7.07(1H,,m),,3.05(1H,,p),2.18(15H,s),,1.20(36H,d);;13C-NMRinCDCl3-d1:δ=152.9,137.7,,130.5,124.7,,27.3,,23.6,,9.7[实施例7]利用Cp*Ti(OMe)3的Cp*Ti(2-苯基苯酚(2-phenylphenolate))3的制备除了代替苯酚而使用2-苯基苯酚(1.12g,6.3毫摩尔)以外,以与上述实施例3相同的方法发生反应并以60%收率获得橙色固体的Cp*Ti(2-苯基苯酚)3(0.83g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R3=R4=R5=R6=氢,R2=苯基),这是通过1H-NMR来确认的。1H-NMRinCDCl3-d1:δ=7.62(3H,,d),7.52(12H,,m),,7.41(3H,,m),7.24(3H,,t),,7.10(6H,,m),,2.18(15H,,s);;13C-NMRinCDCl3-d1:δ=156.2,137.9,,131.2,,130.5,129.0,,127.9,121.8,,116.4,9.57[实施例8]利用Cp*Ti(OMe)3的Cp*Ti(1-萘酚(1-naphtolate))3的制备除了代替苯酚而使用1-萘酚以外,以与上述实施例3相同的方法发生反应来并66%收率获得橙色固体的Cp*Ti(1-萘酚)3(0.81g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R4=R5=R6=氢,R2和R3与1,3-丁二烯基相连接并形成环),这是通过1H-NMR来确认的。1H-NMRinCDCl3-d1:δ=8.22(3H,d),,8.10(3H,d),,7.72(3H,d),,7.61(3H,,m),,7.58(3H,,m),,7.40(3H,t),,6.65(3H,d),,2.18(15H,,s);13C-NMRinCDCl3-d1:δ=151.5,,134.7,,130.5,127.9,,126.8,,126.6,,126.2,123.0,121.0,,109.4,,9.7[实施例9]利用Cp*Ti(OMe)3的Cp*Ti(4-甲基苯酚(4-methylphenolate))3的制备除了代替苯酚而使用4-甲基苯酚(0.69g,6.3毫摩尔)以外,以与上述实施例3相同的方法发生反应并以78%收率获得橙色固体的Cp*Ti(4-甲基苯酚)3(0.78g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R2=R3=R5=R6=氢,R4=甲基),这是通过1H-NMR来确认的。1H-NMRinCDCl3-d1::δ=7.06(6H,d),,6.83(6H,,d),,2.34(9H,s),2.20(15H,s);;13C-NMRinCDCl3-d1:δ=154.3,,131.0,,130.5,,130.4,,115.8,,21.3,,9.7[实施例10]利用Cp*Ti(OiPr)3的Cp*Ti(4-甲氧基苯酚(4-methoxyphenolate))3的制备除了代替苯酚而使用4-甲氧基苯酚(0.77g,6.3毫摩尔)以外,以与上述实施例4相同的方法发生反应并以90%收率获得橙色固体的Cp*Ti(4-甲氧基苯酚)3(0.99g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R2=R3=R5=R6=氢,R4=甲氧基),这是通过1H-NMR来确认的。1H-NMRinCDCl3-d1:δ=6.84(12H,m),3.83(9H,,s),,2.18(15H,,s);13C-NMRinCDCl3-d1:δ=153.2,,149.6,,130.5,,115.7,116.9,55.8,9.7[实施例11]利用Cp*Ti(OiPr)3的Cp*Ti(4-N,N-二甲胺基苯酚(4-N,N-diemthylaminophenolate))3的制备除了代替苯酚而使用4-N,N-二甲胺基苯酚(0.85g,6.3毫摩尔)以外,以与上述实施例4相同的方法发生反应并以85%收率获得橙色固体的Cp*Ti(4-N,N-二甲胺基苯酚)3(1.01g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3、R2=R3=氢,R5=R6=(CH2)4),这是通过1H-NMR来确认的。1H-NMRinCDCl3-d1:δ=6.77(6H,d),6.59(6H,d),,3.06(18H,s),,2.22(15H,,s);13C-NMRinCDCl3-d1:δ=146.8,,143.7,,130.5,116.8,115.7,41.3,9.7[实施例12]利用Cp*Ti(OiPr)3的Cp*Ti(5,6,7,8-四氢-1-萘酚(5,6,7,8-Tetrahydro-1-naphtholate))3的制备除了代替苯酚而使用5,6,7,8-四氢萘酚(0.91g,6.2毫摩尔)以外,以与上述实施例4相同的方法发生反应并以65%收率获得橙色固体的Cp*Ti(5,6,7,8-四氢-1-萘酚)3(0.81g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R4=R5=R6=氢,R2和R3与1、3-丁烯相连接形并成环),这是通过1H-NMR来确认的。1H-NMRinCDCl3-d1:δ=6.70(3H,t),6.48(3H,d),6.40(3H,d),2.74(12H,t),2.21(15H,s),1.72(12H,m);13C-NMRinCDCl3-d1:δ=158,139,131,128,127.1,120.5,113,29.8,23.0,22.7,9.7[实施例13]利用Cp*Ti(OiPr)3的Cp*Ti(4-t-丁基苯酚(4-t-Butylphenolate))3的制备除了代替苯酚而使用4-t-丁基酚(0.91g,6.1毫摩尔)以外,以与上述实施例4相同的方法发生反应并以65%收率获得橙色固体的Cp*Ti(4-t-丁基苯酚)3(1.1g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R2=R3=R5=R6=氢,R4=t-丁基),这是通过1H-NMR来确认的。1H-NMRinCDCl3-d1:δ=7.42(6H,d),6.87(6H,d),2.2(15H,s),1.34(27H,s);13C-NMRinCDCl3-d1:δ=154.2,144.0,131.5,126.4,116.0,31.3,9.8[比较例1]利用Cp*TiCl3的Cp*Ti(4–t–辛基苯酚)3的制备向反应容器投入三氯化镁(五甲基环戊二烯)钛(IV)(Cp*TiCl3,0.578g,1当量)和4-t-辛基苯酚(1.238g,3.1当量)后,投入了甲苯(50mL)。向反应容器连接冷凝器后,回流搅拌12小时。若黄色的反应溶液变成橙色,则以56%收率获得橙色固体的Cp*Ti(4–t–辛基苯酚)3(0.85g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R2=R3=R4=R6=氢,R4=t-辛基),这是通过1H-NMR来确认的。图6示出使上述比较例1的Cp*TiCl和3.1当量的4–t–辛基苯酚发生反应后的生成物Cp*Ti(4–t–辛基苯酚)3的1H-NMR数据。[比较例2]利用Cp*TiCl3/Et3N的Cp*Ti(4–t–辛基苯酚)3的制备在反应容器混合三氯化镁(五甲基环戊二烯)钛(IV)(Cp*TiCl3,0.578g,1当量)和4-t-辛基苯酚(1.238g,3.1当量)后,投入甲苯(50mL)和3.2当量的三乙胺(0.65g)。在混合反应物后,在室温条件下搅拌12小时。若黄色的反应溶液变成橙色,则生成白色的三乙胺盐酸盐。通过过滤去除三乙胺盐酸盐后,通过去除溶液来以59%收率获得橙色固体的Cp*Ti(4–t–辛基苯酚)3(0.89g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R2=R3=R4=R6=氢,R4=t-辛基),这是通过1H-NMR来确认的。图7示出使上述比较例2的Cp*TiCl、3.2当量的三乙胺和3.1当量的4–t–辛基苯酚发生反应后的生成物Cp*Ti(4–t–辛基苯酚)3的1H-NMR数据。[比较例3]利用Cp*TiCl3的Cp*Ti(苯酚)3的制备除了代替4–t–辛基苯酚而使用苯酚(0.583g,3.1当量)以外,以与上述实施例2相同的方法发生反应并以52%收率获得橙色固体的Cp*Ti(苯酚)3(0.48g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R2=R3=R4=R5=R6=氢),这是通过1H-NMR来确认的。[比较例4]利用Cp*TiCl3的Cp*Ti(2,6-二甲基苯酚)3的制备除了代替苯酚而使用2,6-甲基苯酚(0.79g,6.3毫摩尔)以外,以与上述实施例2相同的方法发生反应并以89%收率获得橙色固体的Cp*Ti(2,6-二甲基苯酚)3(0.97g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R3=R5=R4=氢,R2=R6=甲基),这是通过1H-NMR和13C-NMR来确认的。[比较例5]利用Cp*TiCl3的Cp*Ti(2,6-二甲基苯酚)3的制备除了代替苯酚而使用2,6-二异丙基苯酚(1.2g,6.3毫摩尔)以外,以与上述实施例2相同的方法发生反应并以93%收率获得橙色固体的Cp*Ti(2,6-二甲基苯酚)3(1.33g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R3=R5=R4=氢,R2=R6=异丙基),这是通过1H-NMR来确认的。[比较例6]利用Cp*TiCl3的Cp*Ti(2-苯基苯酚(2-phenylphenolate))3的制备除了代替苯酚而使用2-苯基苯酚(1.12g,6.3毫摩尔)以外,与上述实施例2相同的方法发生反应并以55%收率获得橙色固体的Cp*Ti(2-苯基苯酚)3(0.76g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R3=R4=R5=R6=氢,R2=苯基),这是通过1H-NMR来确认的。[比较例7]利用Cp*TiCl3的Cp*Ti(1-萘酚)3的制备除了代替苯酚而使用1-萘酚(0.89g,6.3毫摩尔)以外,以与上述实施例2相同的方法发生反应并以65%收率获得橙色固体的Cp*Ti(1-萘酚)3(0.80g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R4=R5=R6=氢,R2和R3与1,3-丁二烯基相连接并形成环),这是通过1H-NMR来确认的。[比较例8]利用Cp*TiCl3的Cp*Ti(4-甲基苯酚)3的制备除了代替苯酚而使用4-甲基苯酚(0.69g,6.3毫摩尔)以外,以与上述实施例2相同的方法发生反应并以78%收率获得橙色固体的Cp*Ti(4-甲基苯酚)3(0.78g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R2=R3=R5=R6=氢,R4=甲基),这是通过1H-NMR来确认的。[比较例9]利用Cp*TiCl3的Cp*Ti(4-甲氧基苯酚)3的制备除了代替苯酚而使用4-甲氧基苯酚(0.77g,6.3毫摩尔)以外,以与上述实施例2相同的方法发生反应并以78%收率获得橙色固体的Cp*Ti(4-甲氧基苯酚)3(0.99g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R2=R3=R5=R6=氢,R4=甲氧基),这是通过1H-NMR来确认的。[比较例10]利用Cp*TiCl3的Cp*Ti(4-N,N-二甲胺基苯酚)3的制备。除了代替苯酚而使用4-N,N-二甲胺基苯酚(0.85g,6.3毫摩尔),以与上述实施例2相同的方法发生反应并以85%收率获得橙色固体的Cp*Ti(4-N,N-二甲胺基苯酚)3(1.01g)(在化学式1中,M=Ti,Cp=五甲基环戊二烯,n=3,R2=R3=R5=R6=氢,R4=二甲胺基),这是通过1H-NMR来确认的。针对在上述实施例及比较例所制备的生成物,以离子色谱法(Ionchromatography)检测残留氯含量,并记载在下述表1。[表1][实施例13~18及比较例11~12]活性聚合检测使用溶液连续聚合装置如下地进行乙烯和1-辛烯的共重合。在进行充分地干燥后,在由氮取代的1000mL容量的不锈钢连续聚合反应器维持相同的乙烯、1-辛烯、作为铝助催化剂的改良甲基铝氧烷-7(阿克苏·诺贝尔(AkzoNobel)公司,modifiedMAO-7,7wt%的铝Isopar溶液)及作为硼类助催化剂的三苯碳鎓四(五氟苯基)硼酸盐(trityltetrakis(pentafluorophenyl)Borate)的投入量及反应温度(催化剂注入温度、反应器温度),并从将甲基环已烷作为反应溶剂来投入的催化剂的量(μmole/kg)和乙烯转换率比较催化剂的活性。乙烯、1-辛烯及助催化剂的投入量和反应器内催化剂的滞留时间为如下表2。[表2]项目注入量总溶液流量(kg/h)(甲基环已烷)5乙烯投入量(wt%)101-辛烯投入比(C8/C2比)0.19催化剂的反应器内滞留时间(min)8乙烯注入量(g/h)5001-辛烯注入量95铝助催化剂的投入量(μmole/kg)280硼类助催化剂的投入量(μmole/kg)56将在实施例1、7、13及比较例2中分别合成的Cp*Ti(4–t–辛基苯酚)3、Cp*Ti(2-苯基苯酚)3、Cp*Ti(4-t-丁基苯酚)3及Cp*Ti(4–t–辛基苯酚)3分别制备成1mM的甲苯溶液并投入连续聚合反应器后,连续供应至反应器内,并以乙烯实现聚合。调节甲基环已烷溶剂的注入量,以使催化剂的反应器内滞留时间成为8分钟,维持一定的催化剂注入温度和反应器的温度,并检测催化剂注入量。为了检测乙烯转换率,而检测了所生成的高分子的重量,并以针对乙烯注入量的比率进行比较。在表3示出在催化剂投入温度为60℃、反应器温度为150℃的条件下生成的高分子密度、分子量(MI)、转换率及催化剂投入量。[表3]并且,在下述表4示出在催化剂投入温度为70℃、反应器温度为160℃的条件下生成的高分子密度、分子量(MI)、转换率及催化剂投入量。[表4]从表3和表4中可知,在实施例13至实施例14的聚合温度为150℃和160℃条件下的催化剂注入量低于比较例11及比较例12的催化剂化合物。即,可知,在生产等量的高分子的过程中,与借助伴随氯离子的制备方法合成的催化剂化合物相比,借助本发明的排除氯离子的合成方法制备的催化剂化合物的活性实现少量的催化剂投入量。本发明实施例的化合物具有较低的催化剂注入量的特性,即,展示较高的活性聚合,这种现象基于催化剂的制备方法,在比较例的情况下,由于包含氯离子的三乙胺盐酸盐存在于催化剂化合物内,因此会给活性聚合带来影响。尤其,在使用本发明的从过渡金属醇盐前体进行制备并通过升华/过滤提纯的催化剂的实施例13至实施例18的情况下,与使用从现有过渡金属氯化物前体中制备的催化剂的比较例11及比较例12相比,可容易地去除催化剂化合物内的杂质,因此催化剂活性相对较高。并且,在比较例11及比较例12中使用的比较例1的催化剂作为从过渡金属氯化物前体制备的催化剂,来源于起始原料的氯化物残留在催化剂内,因此具有腐蚀在聚合工序使用的反应器等的材质、生成作为杂质的催化剂变形物质以及由于很难注入催化剂而不容易精确地控制聚合反应的缺点。产业上的可利用性本发明的过渡金属配合物的制备方法具有如下的优点,即,作为起始原料,使不包含卤素的过渡金属醇盐前体,尤其不包含氯的过渡金属醇盐前体和可通过升华或单纯过滤来提纯的酚类配体发生反应,从而以高收率制备过渡金属配合物,使成为现有问题的借助水分的变性种的形成最小化,并且可通过升华或单纯过滤提纯简单地分离未与所制备的过渡金属配合物发生反应的酚类配体。并且,在制备过渡金属配合物过程中,由于不包含卤素,尤其完全不包氯,因此即使将所制备的过渡金属配合物利用于烯烃聚合,也不用担心腐蚀工序上的材质,并且在作为往使用的起始原料的过渡金属氯化物前体的情况下,由于必须需要同时使用胺类化合物,因此使生成物和胺残物共存,但在作为起始原料使用过渡金属醇盐前体而不是氯化物的本发明的情况下,具有不使胺残物等的杂质共存的优点。并且,由于反应选择性优秀,具有可以仅通过反应物质的摩尔比变化来简单地制备结合有1、2或3个酚类配体的过渡金属配合物的优点。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1