表面活性剂组合物的制作方法
本发明涉及适用于例如生产水性环氧分散体组合物的表面活性剂组合物。
背景技术:
通常,环氧官能化表面活性剂用于制造水性环氧分散体,其中表面活性剂用于分散环氧树脂,例如酚醛清漆、液体环氧树脂(例如dertm330,可购自陶氏化学公司(thedowchemicalcompany)的液体环氧树脂[ler])和“1-型”环氧树脂。已知的环氧官能化非离子表面活性剂通常利用一个或多个来自上述已知环氧树脂的环氧基团来官能化。例如,一些已知的环氧反应性表面活性剂包括美国专利第5,118,729号、美国专利第4,421,906号、美国专利第5,602,193号和美国专利第6,221,934号中公开的环氧官能性聚醚表面活性剂。
当在分散体中使用上述已知的环氧官能化非离子表面活性剂时,除非在此类分散体中使用额外的溶剂,并且同时仍然保持在合理的分散体粘度范围内(例如,粘度小于[<]约10pa-s),否则只能获得低固体含量(例如,对于ler分散体,小于约65重量百分比(wt%),对于酚醛清漆分散体,小于约60wt%,或者对于1-型分散体,小于约55wt%)。
美国专利第5,118,729号公开了甲氧基-聚乙二醇环氧官能化表面活性剂(单酯)和可用作疏水物的高级树脂。例如,美国专利第5,118,729号公开了der331和den438和单酯,及然后分散的官能化环氧树脂。美国专利第5,118,729号未公开将分散剂的环氧官能团与环氧树脂相匹配,或疏水物的分子量(mw)对分散特性有什么影响。
美国专利第8,580,871号公开了制备环氧官能化表面活性剂的方法,其是通过首先使酰胺基胺与酸封端的聚氧化烯多元醇反应。然后,使环氧官能化表面活性剂与环氧基反应以制造非离子环氧官能化表面活性剂,其进而可用于分散环氧树脂。环氧官能化表面活性剂可进一步与胺化合物反应,使得所得反应产物可用作固化剂。环氧树脂可以是液体或聚合物。美国专利第8,580,871号中列出的潜在环氧树脂候选物包括芳香族、脂肪族和酚醛环氧树脂或其混合物。表面活性剂可以单独或原位制造。美国专利第8,580,871号未公开改变表面活性剂中环氧疏水物的官能度或其对分散性质的影响。美国专利第8,580,871号也未教导降低分散体的粘度或在醇的存在下改善分散体的分散稳定性。表面活性剂可以用分散的树脂原位制造,然而美国专利第8,580,871号未教导将疏水物调整为环氧树脂,也未教导调整疏水物对分散性能的影响。
美国专利第4,423,201号公开了通过使脂肪族聚醚二醇(peg)与二异氰酸酯和二元酚反应来制造环氧官能化表面活性剂的方法。peg的mw在约800至约20,000之间变化。美国专利第4,423,201号公开了将二异氰酸酯的脂肪族或芳香族性质与环氧化物的类似性质相匹配。有用的peg包括含有50wt%至90wt%eo的环氧乙烷(eo)/环氧丙烷(po)嵌段共聚物(其粘度在约5,000mpa-s至约10,000mpa-s的范围内)。美国专利第4,423,201号的方法包括使异氰酸酯与乙二醇反应,直到所有的异氰酸酯基团消失,且然后与二元酚的二缩水甘油基醚(dge)和二元酚(例如双酚a)共反应。美国专利第4,423,201号未公开改变疏水物的mw,或者使疏水物与被分散的树脂匹配。上述专利中公开的实例在表面活性剂中使用液体环氧树脂。
美国专利第5,602,193号公开了环氧官能化聚醚表面活性剂,其允许粒径<1微米的环氧分散体。环氧基对称地连接到eo或eo/po链(a和b)的任一端。两种化合物(a和b)由eo和/或eo/po链组成,在任一侧具有酯基。这些是脂肪族分子。表面活性剂的mw为1,000至40,000;且优选在2,000和20,000之间。美国专利第5,602,193号描述了使氧化的多元醇链上的羧酸与环氧化物基团反应以形成表面活性剂。优选氧化的聚亚烷基二醇(例如eo、po、环氧丁烷[bo]的一些组合)。美国专利第5,602,193号公开了脂肪族或芳香族环氧树脂的使用;并且在合成表面活性剂中使用液体环氧树脂(具有约190的eew)。然而,美国专利第5,602,193号未公开可用作表面活性剂的疏水物的各种不同的树脂,或此类疏水物对分散体或其性能性质的影响。
美国专利第5,648,409号公开了由与一定量的聚亚烷基胺反应的树脂组成的自分散环氧,其中亚烷基部分由eo和/或po基团组成,且mw为约2,000至约10,000(例如jeffaminem-2070),以制造环氧/胺加合物。使用eew小于约400的双酚a基和酚醛清漆型环氧树脂(例如der331)。美国专利第5,648,409号未公开改变表面活性剂的疏水部分,或此类疏水物对分散性质的影响。
美国专利第6,410,617号公开了制造环氧官能化表面活性剂的方法,其通过使羧酸酯(例如马来酸二乙酯)与聚亚烷基二醇胺(例如jeffamine2070)反应,然后添加双酚a和液体环氧树脂并使之反应。所得表面活性剂用丙氧基乙醇稀释。美国专利第6,410,617号未公开改变疏水物mw或某一mw的此类疏水物对分散/性能性质的影响。
技术实现要素:
本发明的一个实施例涉及环氧官能化表面活性剂组合物,其包括以下各项的反应产物:(a)亲水材料和(b)疏水物,其中疏水物是平均mw大于约370道尔顿(da)的环氧树脂,如通过凝胶渗透色谱法(gpc)测量。
本发明的另一个实施例涉及水性环氧分散体组合物,其包括:(a)上述环氧官能化表面活性剂组合物;(b)环氧树脂;和(c)水。
本发明的其它实施例包括制备上述环氧官能化表面活性剂组合物的方法和制备上述水性环氧分散体组合物的方法。
本发明部分地包括发现表面活性剂组合物,其使用平均mw大于370da的疏水物组分(b)(例如der664)制备环氧官能化表面活性剂组合物。然后,当使用分散在环氧树脂中的本发明的上述表面活性剂组合物制备水性环氧分散体组合物时,水性环氧分散体组合物的所得粘度可以低于使用基于较低mw疏水物的表面活性剂的常规水性环氧分散体组合物的粘度。本文讨论了本发明的其它益处、优点和目的。
附图说明
为了说明本发明,附图示出了目前优选的本发明的形式。然而,应当理解,本发明不限于附图中所示的实施例。
图1是示出了使用相对于eew的术语“类型”的各种环氧树脂聚合物的名称以及环氧树脂聚合物主链中的重复基团的n值的示意图。
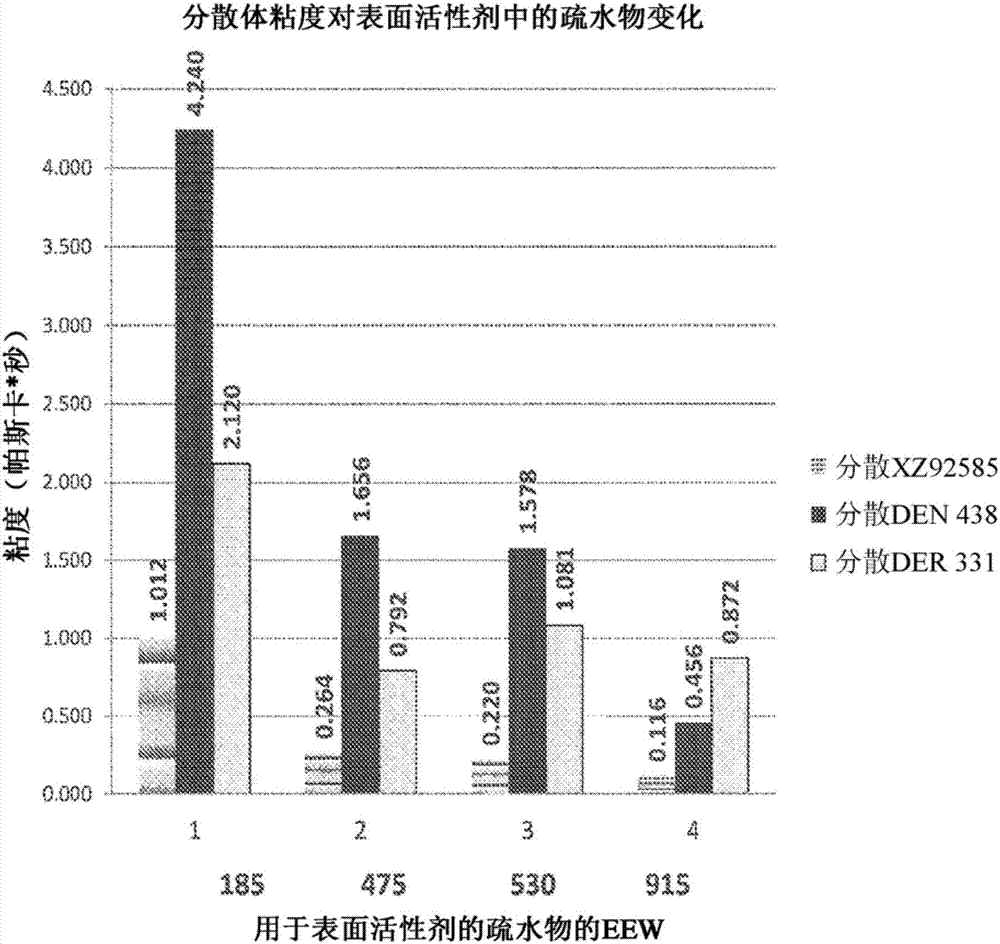
图2是示出了分散体粘度对各种组合物的疏水物mw的图解说明。随着用于制造表面活性剂的疏水物的eew增加,分散体的粘度降低。这在分散1-型树脂、标准商业酚醛清漆树脂如den438和液体环氧树脂如der331时看出。图2所示的所有三种分散体的平均粒径保持在450纳米(nm)和650nm之间。实例1的树脂(1-型树脂)的固体含量保持在55wt%。比较实例a和比较实例b的分散体保持在60wt%固体。
具体实施方式
关于环氧树脂,术语“1-型”、“4-型”和其它“类型”是与本领域公知的环氧树脂相关的术语。这些术语可以与诸如“类型-1”、“类型-2”等的名称互换使用。没有对“1-型”树脂或“2-型”树脂等的构成进行严格定义,然而,本领域技术人员可以遵循图1作为指导,其中图1示出了相对于eew的“类型”树脂和环氧树脂聚合物(例如具有以下结构(i)的化学式的环氧树脂)的主链中的重复基团的n值:
本发明的一个广泛实施例涉及提供环氧官能化表面活性剂组合物,其包括至少以下各项的反应产物:(a)亲水材料和(b)疏水材料(或“疏水化合物”)。
在一个实施例中,用于制备本发明的环氧官能化表面活性剂组合物的亲水材料组分(a)可以包括一种或多种本领域已知的常规亲水材料。例如,亲水材料可以包括烷基葡糖苷、甘油、烷基酯、脂肪醇、烷基二醇、乙氧基化直链醇、乙氧基化烷基酚、脂肪酸酯、胺和酰胺衍生物、环氧乙烷共聚物、环氧丙烷共聚物、多元醇、乙氧基化多元醇、硫醇及其混合物。在一个实施例中,亲水材料通常具有约100至约20,000的平均mw;在另一个实施例中为约500至约10,000;以及在又一个实施例中为约1,400至约8,000。
用于制备本发明的环氧官能化表面活性剂组合物的其它亲水材料组分(a)可以包括(例如):
(i)r-o(ch2-ch2-o)nch2-ch2-oh,其中r可以是甲基、乙基或c3-c20基团,并且n可以是1至452;
(ii)r-o(ch2-ch2-o)n[x]m-oh,其中r和n如上所定义,x可以是ch2-chch3、ch2-ch2-ch2–、ch3-ch2-ch2-ch2-o、c5至c20直链基团,或支链r-o(ch2-ch2-o)n[x]m-(ch2-ch2-o)n-oh,其中r、x和n如上所定义,且m可以为0至474;
(iii)ho(ch2-ch2-o)n-ohho(ch2-ch2-o)n-[x]moh,其中x可以是ch2-chch3、ch2-ch2-ch2–、ch3-ch2-ch2-ch2-o、c5至c20直链基团,或支链ho(ch2-ch2-o)n[x]m-(ch2-ch2-o)n-oh,其中x、n和m如上所定义;
(iv)ho-[x]m–(ch2-ch2-o)n--[x]m'-ohoh,其中x、n和m如上所定义,且m'可以为0至(474-m);及
(v)其混合物。
在一个优选实施例中,亲水材料包括亚烷基二醇,其在一个实施例中平均mw为约500至约20,000,且在另一个实施例中为约1,000至约15,000,且在另一个实施例中为约1,400至约10,000,且在另一个实施例中为约3,000至约8,000,以及在另一个实施例中为约4,000至约5,000。亚烷基二醇可以是(例如)聚乙二醇。
通常,用于制备本发明的环氧官能化表面活性剂组合物的亲水材料组分(a)的量可以在以下范围内:在一个实施例中为约95wt%至约5wt%,在另一个实施例中为约78wt%至约22wt%,在又一个实施例中为45wt%至约22wt%,以及在又另一个实施例中为40wt%至约30wt%。当亲水材料的用量高于95wt%时,表面活性剂过于亲水并迁移到水相,而不是停留在颗粒界面处。当表面活性剂优选水相时,粘度明显高于如果表面活性剂适当地结合到颗粒上的粘度。当亲水材料的用量小于5wt%时,表面活性剂提供不足的空间稳定性;且因此,在不向树脂共混物中添加大量额外的外部表面活性剂情况下,使得分散体稳定储存超过几周变得困难得多。
在上述第一实施例中,环氧官能化表面活性剂组合物包括至少以下各项的反应产物:(a)亲水材料和(b)疏水化合物;且亲水材料组分(a)包含一种、两种或更多种单独的亲水化合物,且疏水材料组分(b)包含一起反应的一种、两种或更多种单独的化合物。
在另一个第二实施例中,用于制备本发明的环氧官能化表面活性剂组合物的亲水材料组分(a)可以包括单独制备的并且与疏水化合物组分(b)分开制备的前体材料或中间材料,然后使亲水材料与疏水化合物反应。在该情况下,可以首先制备作为前体材料或中间产物的亲水材料,且然后与疏水化合物反应。例如,亲水前体材料可以是以下各项的反应产物:(i)亚烷基二醇化合物和(ii)酸酐化合物。
在另一个第三实施例中,环氧官能化表面活性剂组合物可以包括以下各项的反应产物:(i)亚烷基二醇化合物、(ii)酸酐化合物和(iii)疏水化合物。在该原位型实施例中;(i)亚烷基二醇化合物、(ii)酸酐化合物和(iii)疏水化合物共混并反应以形成环氧官能化表面活性剂组合物。
在实施本发明的优选实施例中,使用上述第二实施例,其中在亲水材料与疏水化合物反应之前首先制备亲水材料。如上所述,亲水材料前体可通过使以下各项反应来制备:(i)亚烷基二醇化合物和(ii)酸酐化合物。
用于制备亲水材料(该亲水材料用于制备本发明的表面活性剂组合物)的亚烷基二醇化合物组分(i)可以包括一种或多种亚烷基二醇化合物。例如,亚烷基二醇化合物可以包括聚乙二醇、甲氧基聚乙二醇、聚环氧乙烷、聚氧乙烯及其混合物。在一个优选实施例中,亚烷基二醇化合物包括聚乙二醇。
通常,用于制备本发明的亲水材料的亚烷基二醇化合物的量可以在以下范围内:在一个实施例中为约4wt%至约70wt%,在另一个实施例中为约19wt%至约60wt%,以及在又一个实施例中为25wt%至约50wt%。本发明中使用的亚烷基二醇化合物的量可以随着所使用的不同表面活性剂和不同疏水基团和所使用的不同化学计量比而变化。然而,通常当使用低于约5wt%水平的量的亚烷基二醇化合物时,表面活性剂的量太低而不能产生小粒径的稳定分散体。并且通常,当使用高于约70wt%水平的量的亚烷基二醇化合物时,亲水部分太大,并且变得太可溶于水而不能停留在水和颗粒的界面处。
用于制备亲水材料(其进而用于制备本发明的表面活性剂组合物)的酸酐化合物组分(ii)可以包括任何数量的酸酐化合物,所述酸酐化合物包括(例如)美国专利第5,118,729号中公开的化合物,其通过引用并入本文。
例如,连接基团是能够与聚乙二醇的单烷基醚的伯羟基部分和多羟基烃的多缩水甘油醚的1,2-环氧部分二者反应的化合物的残基。可以使用具有(1)与羟基部分反应的部分和(2)与缩水甘油基部分反应的部分两者的任何化合物。在优选的实施例中,此类化合物是二羧酸或二羧酸的酸酐。优选的二羧酸或二羧酸的酸酐对应于下式(ii)或(iii)之一:
其中z是c1-c20亚烃基部分。在该实施例中,x由式(iv)表示,其中羰基部分与聚(氧乙烯)单烷基醚的残基结合。
优选z是c1-c20亚烷基、c3-c20亚环烷基、c4-c20亚烷基取代的亚环烷基、c亚芳基或c7-c20烷基取代的亚芳基。更优选z是c1-c20亚烷基、c5-c6亚环烷基、c6-c12亚芳基、c7-c20亚烷基取代的亚环烷基或c7-c20烷基取代的亚芳基。在最优选的实施例中,z对应于下式(v)或(vi):
其中r6是c1-c10烷基,且t是0或1。优选地,r6是c1-c3烷基;且最优选甲基。在本发明中有用的优选酸酐包括以下:邻苯二甲酸酐;1,2,5,6-四氢邻苯二甲酸酐;3-甲基六氢邻苯二甲酸酐;3-乙基六氢邻苯二甲酸酐;及其混合物。酸酐优于羧酸,并且更优选脂环族酸酐,因为脂环族酸酐比芳香族酸酐提供了对水解更稳定的产物。3-烷基六氢邻苯二甲酸酐是最优选的,因为它们的高稳定性。在用于本发明的烷基六氢邻苯二甲酸酐中,3-甲基六氢邻苯二甲酸酐是目前最容易获得的。在另一个实施例中,酸酐化合物可以包括琥珀酸酐。
通常本发明中使用的酸酐化合物的量可以随着所使用的不同表面活性剂和不同亲水基团和所使用的不同化学计量比而变化。然而,理想上,当选择酸酐水平时,优选保持亲水组分在约0.8:1至约1.2:1之间的化学计量比。通常,使用1:1的比例得到最佳反应,并且可以有效监测反应的完成。远离1:1操作可产生与疏水物的未知副反应。
通常,当用于制备本发明的环氧官能化表面活性剂组合物时,包含(i)亚烷基二醇化合物和(ii)酸酐化合物的反应产物的亲水材料的量可以在以下范围内:在一个实施例中为约5wt%至约95wt%,在另一个实施例中为约10wt%至约80wt%,以及在又一个实施例中为20wt%至约40wt%。本发明中使用的亲水材料的量可以随着所使用的不同表面活性剂和不同疏水基团和所使用的不同化学计量比而变化。然而,通常当使用低于约5wt%的水平时,亲水链长度的量太低而不能产生表面活性材料。并且通常,当使用高于约95wt%的水平时,表面活性剂的水溶性使得材料更可能停留在水相中,而不是在颗粒的表面。
用于制备本发明的环氧官能化表面活性剂组合物的疏水物组分(b)可以包括各种环氧化合物。用于制备本发明的环氧官能化表面活性剂组合物的环氧树脂疏水物可以包括一种环氧树脂或两种或更多种环氧树脂的混合物。例如,通常,环氧树脂疏水物可以包括选自脂肪族化合物、脂环族化合物、芳香族化合物、杂环化合物及其混合物的环氧化合物或聚环氧化物。在一个实施例中,环氧化合物可以平均含有一个或多个反应性环氧乙烷基团。用于本文所述的实施例中的环氧树脂可以包括(例如)单官能环氧树脂、多官能环氧树脂或聚官能环氧树脂及其组合。用作本发明的环氧树脂疏水物的环氧树脂和此类环氧树脂的制备公开在(例如)lee,h.和neville,k.,《环氧树脂手册(handbookofepoxyresins)》,麦格劳-希尔图书公司(mcgraw-hillbookcompany),纽约,1967,第2章,第2-1页至第2-27页中,通过引用并入本文。
在本文公开的实施例中使用、用于本发明的疏水组分(b)的环氧树脂可以变化,并且包括常规的和市售的环氧树脂。本文中使用的环氧树脂组分可以包括单独使用的单一环氧树脂化合物或者组合使用的两种或更多种环氧化合物的混合物。环氧树脂(也称为聚环氧化物)可以是每分子平均具有多于一个未反应的环氧化物单元的产物。在选择环氧树脂用于本文公开的疏水物时,应考虑最终产物的性质和粘度以及其它可能影响表面活性剂组合物加工的性质。
用于本发明的合适的常规环氧树脂化合物可以通过本领域已知的方法制备,例如基于表卤代醇与(1)酚或酚型化合物、(2)胺或(3)羧酸反应的反应产物。本文使用的合适的常规环氧树脂也可以由不饱和化合物的氧化制备。例如,本文使用的环氧树脂可以包括表氯醇与多官能醇、酚、双酚、卤化双酚、氢化双酚、酚醛清漆树脂、邻甲酚酚醛清漆、苯酚酚醛清漆、聚乙二醇、聚亚烷基二醇、脂环族、羧酸、芳香胺、氨基酚或其组合的反应产物。环氧化合物的制备描述于(例如)《柯克·奥思默化工百科全书(kirk-othmer,encyclopediaofchemicaltechnology)》,第3版,第9卷,第267-289页中。
在一个实施例中,用于与表卤代醇反应制备环氧树脂的合适的酚、酚型或多元酚化合物包括(例如)每分子平均具有多于一个芳香族羟基的多元酚化合物,例如二羟基酚;双酚;双酚如双酚a、双酚ap(1,1-双(4-羟基苯基)-1-苯基乙烷)、双酚f或双酚k;卤化双酚如四甲基-四溴双酚或四甲基三溴双酚;卤化双酚如四溴双酚a或四氯双酚a;烷基化双酚如四甲基双酚;烷基化双酚;三酚;酚-醛酚醛清漆树脂(即酚和简单醛、优选甲醛的反应产物)如酚-甲醛酚醛清漆树脂、烷基取代的酚-甲醛树脂、酚-羟基苯甲醛树脂,烷基化的酚-羟基苯甲醛树脂或甲酚-羟基苯甲醛树脂;卤化酚-醛酚醛清漆树脂;取代的酚-醛酚醛清漆树脂;酚-烃树脂;取代的酚-烃树脂;烃-酚树脂;烃-卤化酚树脂;烃-烷基化酚树脂;间苯二酚;儿茶酚;氢醌;二环戊二烯-酚树脂;二环戊二烯取代的酚树脂;或其组合。
在另一个实施例中,用于与表卤代醇反应制备环氧树脂的合适的胺包括(例如)二氨基二苯基甲烷、氨基酚、二甲苯二胺、苯胺或其组合。
在另一个实施例中,用于与表卤代醇反应制备环氧树脂的合适的羧酸包括(例如)邻苯二甲酸、间苯二甲酸、对苯二甲酸、四氢和/或六氢邻苯二甲酸、内亚甲基四氢邻苯二甲酸、间苯二甲酸、甲基六氢邻苯二甲酸或其组合。
用于本发明的环氧树脂的几个非限制性实施例包括(例如)由表卤代醇和聚乙二醇反应制备的脂肪族环氧化物,如三甲基丙烷环氧化物;二缩水甘油基-1,2-环己烷二羧酸酯或其混合物;双酚a的二缩水甘油基醚;双酚f的二缩水甘油基醚;间苯二酚二缩水甘油基醚;对氨基酚的三缩水甘油基醚;含卤素(例如氯或溴)的环氧树脂,例如四溴双酚a的二缩水甘油基醚;环氧化酚的酚醛清漆;环氧化双酚a的酚醛清漆;恶唑烷酮改性环氧树脂;环氧封端的聚恶唑烷酮;及其混合物。
用于本发明的合适的市售环氧树脂化合物可以是(例如)可购自陶氏化学公司的环氧树脂,例如环氧树脂的d.e.r.tm300系列、d.e.n.tm400系列、d.e.r.tm500系列、d.e.r.tm600系列及d.e.r.tm700系列。用于本发明的双酚a基环氧树脂的实例包括市售树脂,例如d.e.r.tm300系列和d.e.r.tm600系列,可购自陶氏化学公司。用于本发明的环氧酚醛清漆树脂的实例包括市售树脂,例如d.e.n.tm400系列,可购自陶氏化学公司。
例如,作为本发明的一个说明性实施例,环氧树脂可以是液体环氧树脂,例如d.e.r.383,即环氧当量为约175至约185、粘度为约9.5pa-s且密度为约1.16克/立方厘米的双酚a的二缩水甘油基醚(dgebpa)。可用于环氧树脂组分的其它市售环氧树脂可以是(例如)d.e.r.330、d.e.r.354、d.e.r.332或其组合。
用作组分(b)的其它合适的环氧树脂公开在(例如)美国专利第3,018,262号;第7,163,973号;第6,887,574号;第6,632,893号;第6,242,083号;第7,037,958号;第6,572,971号;第6,153,719号;及第5,405,688号;pct公开wo2006/052727;美国专利申请公开第20060293172号及第20050171237号,每一者在此以引用的方式并入本文。适用于本发明的环氧树脂及其前体的实例还描述于(例如)美国专利第5,137,990号及第6,451,898号,其以引用方式并入本文。
可用于本发明的环氧树脂疏水物的优选实施例包括双酚a的二缩水甘油基醚、酚醛清漆、1-型环氧树脂及其混合物。
在另一个优选的实施例中,根据本发明可使用的市售环氧树脂(可购自陶氏化学公司)可以包括(例如)dertm330、dertm331、dertm661、dertm664、dertm667、dentm438、dertm354及其混合物。
通常,用于制备本发明的环氧官能化表面活性剂组合物的环氧树脂的量是由亲水基团的官能度和亲水基团对疏水基团的化学计量比确定。在一个实施例中,以组合物的树脂形成组分的总重量计,环氧树脂的量可以在约10wt%至约85wt%的范围内,在另一个实施例中为约20wt%至约75wt%,以及在又一个实施例中为40wt%至约65wt%。本发明中使用的环氧树脂的量可以随着所使用的不同表面活性剂和不同疏水基团和所使用的不同化学计量比而变化。然而,通常当使用低于约10wt%的水平时,表面活性剂太可溶于水而不能保持表面活性。并且通常当使用高于约85wt%的水平时,空间稳定性太小而不能具有有效的表面活性剂。
重要的是,用于本发明的环氧树脂疏水物具有优选的mw,使得本发明的环氧官能化表面活性剂组合物和本发明的水性环氧分散体组合物表现出本发明预期的性能性质。通常,环氧树脂疏水物的mw应大于或等于使用由环氧树脂疏水物制备的环氧官能化表面活性剂组合物分散的树脂。例如,在一个实施例中,环氧树脂疏水物的mw通常可以大于约370da;在另一个实施例中为约370da至约4,000da,在又一个实施例中为约800da至约4,000da,以及在又另一个实施例中为约900da至约2,000da。
优选选择亲水材料组分(a)和环氧树脂疏水物组分(b)使得亲水材料和环氧树脂疏水物都不自聚合以产生mw大于上述mw的分子;或产生mw大于上述mw的环氧官能化表面活性剂组合物产物。
通常,在一个实施例中,本发明的环氧官能化表面活性剂组合物的mw可以大于约1,500da;在另一个实施例中为约3,000da至约16,000da,在又一个实施例中为约4000da至约12,000da,以及在又另一个实施例中为约5,000da至约10,000da。
在优选的实施例中,在本发明的实践中可以任选地使用一种或多种合适的反应催化剂。用于制备本发明组合物的催化剂可以选自(例如)金属盐(例如碱金属盐或碱土金属盐)、叔胺、季铵盐、季鏻盐、膦等中的一种或多种,及其混合物。优选地,用于本发明的催化剂是乙基三苯基乙酸鏻、任何脂肪族或芳香族取代的苯基溴化鏻或其混合物。
任选的反应催化剂通常以0wt%至约10wt%、约0.01wt%至约10wt%;优选约0.05wt%至约5wt%、最优选约0.1wt%至约4wt%的量使用,重量百分比以所使用的单体化合物的组合重量计。
制造环氧官能化表面活性剂组合物的方法包括在反应条件下使上述亲水材料和上述环氧树脂疏水物反应的步骤,使得形成环氧官能化表面活性剂组合物。例如,在一个实施例中,用于制备表面活性剂的方法可以是如wo9110695中描述的方法,通过引用并入本文。
在另一个实施例中,实施形成环氧官能化表面活性剂组合物的反应,使得含有环氧官能化表面活性剂组合物产物的反应混合物中的残余酸水平(例如)在合成过程结束时为零或非常接近于零。通常,本发明的环氧官能化表面活性剂组合物中的残余酸水平在一个实施例中可以小于约0.1wt%、在另一个实施例中小于约0.01wt%、以及在又一个实施例中小于约0.001wt%。如果残余酸水平处于高于约0.1wt%的较高水平,则peg/环氧/酸酐可能不转化为所需产物。在合成结束后未与环氧树脂完全反应的游离酸酯将继续缓慢反应或作为增稠剂保留在水相中,这是不希望的。
如上所述,在另一个实施例中,实施反应使得亲水材料和环氧树脂疏水物不自聚合以产生高分子量物质。例如,所得反应产物优选具有小于约500,000da、更优选小于约100,000da以及最优选小于约70,000da的mw。
用于制备环氧官能化表面活性剂组合物的所有组分通常在能够制备有效的表面活性剂组合物的温度下混合和分散。例如,在一个实施例中,所有组分的混合期间的温度通常可以为约40℃至约160℃,且在另一个实施例中为约80℃至约150℃,以及在又一个实施例中为约120℃至约140℃。
本发明的环氧官能化表面活性剂组合物的制备和/或其任何步骤可以是间歇或连续方法。在该方法中使用的混合设备可以是本领域技术人员公知的任何容器和辅助设备。
本发明的环氧官能化表面活性剂组合物的一些有益性质或特性包括(例如):(1)各种环氧树脂疏水物可用于制造表面活性剂组合物,(2)几种环氧树脂疏水物可用于确保树脂/表面活性剂相容性,(3)几种环氧树脂疏水物可用于调节特定水性环氧分散体组合物的粘度,(4)几种亲水物可用于在给定温度下调节表面活性剂组合物的所需表面活性,(5)表面活性剂组合物相对容易大规模生产,和(6)表面活性剂组合物相对容易溶解在被分散的树脂中。
本发明的另一个广泛实施例涉及提供水性环氧分散体组合物,其至少包括:(a)如上所述的环氧官能化表面活性剂组合物;(b)环氧树脂;(c)水和任选地(d)外部表面活性剂。
用于制备本发明的水性环氧分散体组合物的环氧官能化表面活性剂组合物组分(a)可以包括一种或多种如上所述的环氧官能化表面活性剂组合物。
通常,用于制备本发明的水性环氧分散体组合物的环氧官能化表面活性剂组合物组分(a)的量可以在以下范围内:在一个实施例中约0.1wt%至约20wt%,在另一个实施例中约0.5wt%至约15wt%,以及在又一个实施例中1wt%至约10wt%。当所用的环氧官能化表面活性剂组合物的量为小于约0.1wt%水平时,没有足够的表面活性剂来制造小粒径或稳定的分散体。当所用的环氧官能化表面活性剂组合物的量高于约20wt%水平时,使用表面活性剂的分散体容易分散并显示出极好的储存稳定性,但是如果表面活性剂含量太高,则来自表面活性剂的水敏感性将有害地影响水性环氧分散体组合物的性能性质。
用于制备本发明的水性环氧分散体组合物的环氧树脂组分(b)可以包括与用于制备环氧官能化表面活性剂组合物的环氧树脂疏水物有关的上述一种或多种环氧化合物。例如,公开在(例如)通过引用并入本文的lee,h.和neville,k.,《环氧树脂手册(handbookofepoxyresins)》,麦格劳-希尔图书公司,纽约,1967,第2章,第2-1页至第2-27页中的任何上述环氧树脂可以在本文中用作本发明的环氧树脂分散体组合物的环氧树脂组分(b)。用于制备本发明的水性环氧分散体组合物的环氧树脂组分(b)可以与上述环氧树脂疏水物相同或不同。
用于水性环氧分散体组合物的环氧树脂的优选实施例包括(例如)双酚a的二缩水甘油基醚、酚醛清漆、1-型环氧树脂及其混合物。
在另一个优选的实施例中,可用作用于制备本发明的水性环氧分散体组合物的环氧树脂的市售环氧树脂(可购自陶氏化学公司)可以包括(例如)dertm330、dertm331、dertm661、dertm664、dertm667、dentm438、dertm354及其混合物。用于本发明的其它环氧树脂可以包括(例如)wo9110695中所述的环氧树脂,其通过引用并入本文。
通常,用于制备本发明的水性环氧分散体组合物的环氧树脂的量以组合物的树脂形成组分的总重量计,在一个实施例中可以为约5wt%至约70wt%,在另一个实施例中为约40wt%至约70wt%,以及在又一个实施例中为55wt%至约70wt%。通常,用于制备水性环氧分散体组合物中的环氧树脂的量低于约5wt%是无用产物,这是由于环氧含量低。并且通常,当用于制备水性环氧分散体组合物的环氧树脂的量以高于约70wt%的水平使用时,所得产物具有太高的分散体粘度并且通常储存不稳定。
水组分(c),用于制备本发明的水性环氧分散体组合物。水可以是蒸馏水、自来水或其它水源。
通常,本发明中所用的水量可以在以下范围内:在一个实施例中为约30wt%至约95wt%,在另一个实施例中为约30wt%至约60wt%,以及在又一个实施例中为约30wt%至约45wt%。
表面活性剂组分(d)可任选地用于制备本发明的水性环氧分散体组合物。表面活性剂优选非反应性表面活性剂。可用于本发明组合物中的各种表面活性剂可以包括(例如)任何常规的阴离子、阳离子和非离子表面活性剂或其组合。
阳离子表面活性剂的实例包括但不限于具有各种取代或未取代的烃基链或取代或未取代的杂链(例如取代或未取代的烃基链长度,例如约c8至c22)的阳离子表面活性剂。典型的链是烷基、烷氧基烷基、烷基芳基或烷基酰胺基烷基。可用于本发明组合物的其它阳离子表面活性剂包括铵表面活性剂、取代的铵表面活性剂(如烷基取代的铵表面活性剂)、季铵表面活性剂(例如arquadstm)、吡啶鎓表面活性剂、或取代的吡啶鎓表面活性剂(如烷基取代的吡啶鎓表面活性剂);及其混合物。
可用于本发明的非离子和阴离子表面活性剂如美国专利第6,271,287号中所述,其通过引用并入本文。非离子表面活性剂的实例包括但不限于商业非离子表面活性剂,例如hydropalat3037(可得自汉高(henkel))、emulginprt100(可得自汉高)、emulponel42(可得自witco)、disponilta430(可得自汉高)、sorbanoxao(可得自witco)、atsurf108(可得自ici)、pluronicf108(可得自巴斯夫公司(basfcorp.))、emulginprt200(可得自汉高)及其混合物。合适的非离子表面活性剂的其它实例包括乙氧基化单或二烷基酚,例如聚乙二醇壬基或二壬基苯基醚。市售的乙氧基化二烷基苯基醚的实例是igepaldm970二壬基苯基醚(可得自罗纳普朗克(rhonepoulenc))。
阴离子表面活性剂的实例包括但不限于阴离子表面活性剂,例如长链烷基碱金属磺基琥珀酸盐如二辛基磺基琥珀酸钠(例如,aerosolot75,可购自氰胺(cyanamid))、月桂基硫酸钠、具有聚乙二醇十二烷基醚二钠盐的磺基琥珀酸-4酯(例如可购自氰特(cytec)的aerosola102)、烷基二磺化二苯醚二钠盐如单和二烷基二磺化二苯醚二钠盐(例如可购自陶氏化学公司的dowfax2a1)、二己基磺基琥珀酸钠(例如可购自氰胺的aerosolma80)、聚氧-1,2乙二基-ot-十三烷基-uu-羟基磷酸盐(例如可购自罗纳普朗克的rhodofacrs610)、烷基醚硫酸钠盐(例如可购自汉高的disponilfes61或disponilfes993)及其混合物。
通常,当非反应性表面活性剂用于本发明时,其量可以在以下范围内:在一个实施例中为0wt%至约10wt%,在另一个实施例中为约0.01wt%至约3wt%,以及在另一个实施例中为约0.1wt%至约2wt%。如果表面活性剂含量太高,则来自非反应性表面活性剂的水敏感性将有害地影响水性环氧分散体组合物的性能性质。
制造水性环氧分散体组合物的方法包括在反应条件下将上述环氧官能化表面活性剂组合物、上述环氧树脂、水和所需任何其它任选的添加剂共混或混合的步骤,使得形成水性环氧分散体组合物。
用于制备水性环氧分散体组合物的所有组分通常在能够制备有效的水性环氧分散体组合物的温度下混合和分散。例如,在一个实施例中,组分混合以形成水性环氧分散体组合物期间的温度通常可以为约20℃至约170℃,在另一个实施例中为约40℃至约100℃,以及在又一个实施例中为约60℃至约90℃。
本发明的水性环氧分散体组合物的制备和/或其任何步骤可以是间歇或连续方法。在该方法中使用的混合设备可以是本领域技术人员公知的任何容器和辅助设备。
重要的是在如上所述的分散体组合物中提供必需的组分,否则太多的填料落在上述范围之外可导致不稳定的分散体组合物,以及提供在最终涂料制剂中将无法良好共混的分散体组合物。也重要的是在如上所述的分散体组合物中提供必需的组分,否则太少的填料落在上述范围之外可导致无法提供有效分散体组合物的分散体组合物。
本发明的水性环氧分散体组合物表现出使水性环氧分散体组合物可用于各种最终用途应用的几个性能性质。例如,本发明的水性环氧分散体组合物的有益性质或特性之一可以包括低粘度。例如,在一个实施例中,本发明的水性环氧分散体组合物的粘度可以为约20pa·s;在另一个实施例中为约0.05pa·s至约20pa·s,在又一个实施例中为约0.05pa·s至约10pa·s,以及在又另一个实施例中为约0.50pa·s至约5pa·s。
在一个实施例中,当使用分散在环氧树脂中的本发明的上述环氧官能化表面活性剂组合物制备水性环氧分散体组合物时,本发明的水性环氧分散体组合物的所得粘度通常可比当使用常规环氧官能化表面活性剂时的常规水性环氧分散体组合物的粘度低约2倍至约20倍;在另一个实施例中约2倍至约15倍,以及在又一个实施例中约5倍至约10倍。
作为本发明的水性环氧分散体组合物具有低粘度的结果,与通常具有小于55wt%固体含量的已知分散体相比,水性环氧分散体组合物能够包括高固体含量(例如>65wt%)。随着分散体的固体含量增加,分散体的粘度通常也增加到可用的粘度水平(例如,>15pa·s)。在一个实施例中,本发明的水性环氧分散体组合物的固体含量通常高于约65wt%,在另一个实施例中为约40wt%至约65wt%,以及在又一个实施例中为约55wt%至约65wt%。
由该表面活性剂实现的高固体水性环氧分散体组合物的益处是配制者可以在涂料制剂中使用较少的水以实现所需的粘度。这意味着涂料制剂将提供较高的成膜(filmbuild),这是因为较高的固体含量。此外,本实施例的较高固体含量允许配制者有更大的自由包括其它组分(其可以在较低的固体含量下),并且仍然达到必要的固体/粘度范围。
在本发明的另一个实施例中,可以降低或最小化在水性环氧树脂分散体组合物中的表面活性剂荷载,同时可以保持或改善水性环氧分散体组合物的性能性质。例如,用于水性环氧分散体组合物中的本发明的上述环氧官能化表面活性剂组合物的量可以降低至低于本领域目前使用的量的表面活性剂的水平;并且然而,本发明的水性环氧分散体组合物仍然可以表现出良好的性质,例如储存稳定性。
例如,通常水性环氧树脂分散体组合物中的表面活性剂荷载可以为(例如):在一个实施例中约1wt%至约15wt%,在另一个实施例中约1.5wt%至约10wt%,以及在又一个实施例中3wt%至约6wt%。
本发明水性环氧分散体组合物的另一个益处是水性环氧分散体组合物在醇存在下的分散稳定性。例如,当与醇共混时,水性环氧分散体组合物表现出显著提高的分散稳定性。该分散稳定性性质是重要的,因为分散体的许多最终用途应用,例如当用于货物集装箱涂料的富锌底漆中时,涂料组合物需要在配制和施用期间将大量的醇添加到分散体中。在大多数情况下,醇将通过将表面活性剂溶解到水/醇基质中而破坏涂料组合物的稳定性。使用mw增加的疏水物,表面活性剂在水/醇共混物中的溶解度显著降低,且因此不会破坏稳定性。水性环氧分散体组合物在醇中的稳定性增加的性质还改善了配制油漆的冻融稳定性。
本发明水性环氧分散体组合物的另一个益处是,在制备分散体中,操作者可以灵活地使用各种不同分子量和粘度的不同环氧树脂来制备上述环氧官能化表面活性剂组合物的疏水组分,用于水性环氧分散体组合物中;并且仍然实现本发明的水性环氧分散体组合物的粘度降低。例如,将(例如)1-型环氧用于疏水组分(该疏水组分用于制备上述环氧官能化表面活性剂组合物),可以实现由水性环氧分散体表现出的粘度降低。如上所述的疏水基团的调整允许操作者使用可能与所需疏水物最相容的表面活性剂,这又改善了表面活性剂的功效。
在另一个实施例中,水性环氧分散体表现出的粘度降低也可以通过将与更高类型的环氧共混的1-型环氧用于疏水组分来制备上述环氧官能化表面活性剂组合物来实现,所述更高类型的环氧例如为4-型环氧、7-型环氧或其混合物。
在一个实施例中,环氧官能化表面活性剂的环氧树脂疏水物具有大于或等于被分散树脂的mw。例如在本实施例中,被分散的树脂可以是1-型树脂;且用于环氧官能化表面活性剂中的疏水物可以是(例如)1-型树脂。在另一个实施例中,环氧官能化表面活性剂的环氧树脂疏水物的mw小于被分散树脂的mw,但是在所有情况下大于或等于1-型环氧的mw。
作为一个说明但不限于此,疏水物的mw在一个实施例中通常可为约385da至约4,000da,在另一个实施例中为约500da至约2,000da,以及在又一个实施例中为约700da至约1,900da。相应地,被分散树脂的mw通常可以为约370da至约4,000da。
以下实例和比较实例进一步详细说明本发明,但不应解释为限制其范围。除非另有说明,否则所有份数和百分比均以重量计。
在实例中使用以下标准分析设备和方法:
粒径
使用ls13320贝克曼库尔特(beckmancoulter),环氧型sop进行粒径分析。将少量分散体(约
eew
将metrohm801_1roboticusb样品处理器xl和两个样品的800dosino用于试剂来测量eew(环氧当量)。使用的试剂是0.10n高氯酸的乙酸溶液和四乙基溴化铵。用于分析的电极是854iconnect。对于每个样品,将1g分散体称重到塑料样品杯中。然后首先添加30mlthf(四氢呋喃)并混合1分钟(min)以破坏分散体上的壳。接下来,添加32ml冰醋酸并再混合1分钟以完全溶解样品。然后将样品置于自动取样器上,并将所有相关数据(例如样品id、样品重量)添加到软件。从这里点击开始按钮以开始滴定。此后,添加15ml四乙基溴化铵,且然后缓慢添加高氯酸直到达到电位终点。一旦达到电位滴定终点,软件基于所使用的样品和高氯酸的量计算eew值。
粘度
粘度使用brookfieldrvdv-iprime粘度计测量。将样品倒入100ml玻璃广口瓶中。使用10转/分钟(rpm)的速度,使用适当的心轴(#2-#7)测量粘度,其给出读数为15%-85%的扭矩,以获得最佳可能的结果。当扭矩%和粘度读数都稳定约30秒(s)时,分析完成。
酸百分比
使用25ml滴定管、磁力搅拌板、磁力搅拌棒、flacktekdac150.1fvz-k高速混合器、氢氧化钠溶液n/10、丙酮和酚酞的异丙醇溶液测量酸百分比(酸%)。将样品称重至100最大速度混合器杯中,约4g,并添加丙酮(足以填充速度混合器杯3/4满)作为载体流体。将样品和丙酮置于3500rpm的速度混合器中4分钟;较高mw样品可能需要更多的混合时间。一旦速度混合,将4滴酚酞的异丙醇溶液作为颜色指示剂添加到样品中。将小搅拌棒置于样品杯中,并将样品杯置于磁力搅拌器上。低速搅拌样品杯的内容物。然后将氢氧化钠溶液添加到杯内容物中直至达到颜色终点。使用样品重量和所用氢氧化钠溶液的量(以ml计),如下计算酸%:
酸%=(滴定剂mlx4.502)/(样品重量(g)x10)
上述方程是计算酸%的原始方程的简化版本。原始方程如下:
酸%[wt%]=n*f*45.02*(v2-v1)/10*w
其中n=naoh滴定试剂的理论当量浓度[摩尔/l],
f=当量浓度校正因子,
45.02=酸官能团(cooh)的分子量[g/摩尔],
v1=试剂的空白体积[ml],
v2=样品滴定中所需的试剂体积[ml],且
w=样品质量[g]。
分子量(mw)
通过常规尺寸排阻色谱法(sec)分析样品。结果是基于用线性聚苯乙烯标准物的校准。使用以下校准:
样品制备:2mg/ml在n,n-二甲基甲酰胺(dmf)中+硝酸锂,4g/l;溶液在注射前通过0.45μm尼龙注射器过滤器过滤。
泵:watersmodel2690,标称流速为1.0ml/min。
洗脱液:赛默飞世尔(fisherscientific),n,n二甲基甲酰胺+硝酸锂,4g/l。
注射器:watersmodel2690设置为注射50μl。
柱:两个polymerlabs5μm混合型d柱,批次39.32、57.50,保持在35℃。
检测:含有不同折射率的shodexri-201和waters双波长uv检测器,波长设置为280nm。
数据系统:polymerlabscirrusv.3.3。
校准:来自polymerlabs的10个窄的聚环氧乙烷(peo)标准物,符合一阶多项式曲线。
表面活性剂实例1-使用eew为185的疏水物合成表面活性剂
本发明的环氧官能化表面活性剂组合物是通过根据以下一般程序使亲水部分和环氧疏水物反应而制备:
在该实例中使用1l三颈圆底玻璃反应器。在整个操作过程中,在反应器中保持干燥氮气的气氛。将165.88gmw为4,600的聚乙二醇(peg)装填入反应器中。peg是在室温(rt)下的固体添加物并且在机械搅拌下加热。将搅拌的材料以0.8℃/分钟(最大能力)的速率加热至140℃的温度。在140±3℃下,向混合物中添加19.21g的2-十二碳烯-1-基琥珀酸酐。混合物的温度保持在低于145℃。从反应器中取出反应混合物的样品用于酸含量滴定(酸%:目标1.76,范围1.76至2.10:通常在1l规模下为3.5小时)。
将64.91g的环氧疏水物der330添加到反应器中(在室温下滴加环氧)。将反应器内容物加热至140℃。在140±-3℃下,将0.1g的a1催化剂(乙基三苯基乙酸鏻)添加到反应器材料中。将反应器材料保持在绝热条件下以达到150℃的峰值放热温度。在放热后约30分钟(min),从反应器中取出2.0g样品用于酸滴定测量,并跟踪反应器材料的酸值,直到
将制造的表面活性剂称为环氧表面活性剂“e-peg4600-330”,以标识环氧-peg反应产物;并标识使用mw为4600的peg和der330环氧形成了产物。
表面活性剂实例2-使用eew为485的疏水物合成表面活性剂
本发明的环氧官能化表面活性剂组合物与表面活性剂实例1类似地制备,除了将46.67gmw为4,600的peg装填到反应器中并在反应的第一步骤中添加5.41g的2-十二碳烯-1-基琥珀酸酐。在反应的第二步骤中将47.92g环氧疏水物实验1-型xz92585树脂(eew485)添加到反应器中。
将制造的表面活性剂称为环氧表面活性剂“e-peg4600-585”,以标识环氧-peg反应产物;并标识使用mw为4600的peg和xz92585环氧形成了产物。
表面活性剂实例3-使用eew为530的疏水物合成表面活性剂
本发明的环氧官能化表面活性剂组合物与表面活性剂实例1类似地制备,除了将44.30gmw为4,600的peg装填到反应器中并在反应的第一步骤中添加5.13g的2-十二碳烯-1-基琥珀酸酐。在反应的第二步骤中将50.56g环氧疏水物der661树脂(eew530)添加到反应器中。
将制造的表面活性剂称为环氧表面活性剂“e-peg4600-661”,以标识环氧-peg反应产物;并标识使用mw为4600的peg和der661环氧形成了产物。
表面活性剂实例4-使用eew为915的疏水物合成表面活性剂
本发明的环氧官能化表面活性剂组合物与表面活性剂实例1类似地制备,除了将32.43gmw为4,600的peg装填到反应器中并在反应的第一步骤中添加3.76g的2-十二碳烯-1-基琥珀酸酐。在反应的第二步骤中将63.81g环氧疏水物der664树脂(eew915)添加到反应器中。
将制造的表面活性剂称为环氧表面活性剂“e-peg4600-664”,以标识环氧-peg反应产物;并标识使用mw为4600的peg和der664环氧形成了产物。
分散体实例5-使用在表面活性剂实例1中制备的表面活性剂制备水性环氧分散体
将9.7kg批量的der331和0.3kg表面活性剂e-peg4600-330在夹套反应器中在95℃预共混。一旦均匀,将材料冷却至80℃。然后将冷却的预共混物材料以60g/min泵送到转子定子混合器中。将e-sperse100阴离子表面活性剂(60%活性)流以1.75g/min泵送到混合器中。水也以10g/min添加到混合器中。
然后用额外的水稀释所得高内相乳液以获得58.05%的固体分散体。所获得分散体的平均粒径为~500nm,其中90%的总颗粒低于800nm(“d90%截止点”)。所得分散体的粘度为1.256pa-s。
分散体实例6-使用在表面活性剂实例1中制备的表面活性剂制备水性环氧分散体
类似于分散体实例5实施分散体实例6,除了将9.7kg的den438和0.3kg表面活性剂e-peg4600-330在夹套反应器中在95℃预共混。
所得
分散体实例7-使用在表面活性剂实例1中制备的表面活性剂制备水性环氧分散体
类似于分散体实例5实施分散体实例7,除了将9.5kg的实验1-型xz92585和0.5kg表面活性剂e-peg4600-330在夹套反应器中在95℃预共混。
所得
分散体实例8-使用在表面活性剂实例1中制备的表面活性剂制备水性环氧分散体
类似于分散体实例5实施分散体实例8,除了将der667(80%)、der330(5%)和propylcellosolv(15%)(可购自陶氏化学公司)的预共混物9.02kg与0.98kg表面活性剂e-peg4600-330在夹套反应器中在95℃预共混。
所得
分散体实例9-使用在表面活性剂实例2中制备的表面活性剂制备水性环氧分散体
将9.7kg批量的der331和0.3kg表面活性剂e-peg4600-585在夹套反应器中在95℃预共混。一旦均匀,将材料冷却至80℃。然后将冷却的预共混物材料以60g/min泵送到转子定子混合器中。将e-sperse100阴离子表面活性剂(60%活性)流以1.75g/min泵送到混合器中。水也以10g/min添加到混合器中。
然后用额外的水稀释所得高内相乳液以获得59.94%的固体分散体。所获得
分散体实例10-使用在表面活性剂实例2中制备的表面活性剂制备水性环氧分散体
类似于分散体实例5实施分散体实例10,除了将9.7kg的den438和0.3kg表面活性剂e-peg4600-585在夹套反应器中在95℃预共混。
所得
分散体实例11-使用在表面活性剂实例2中制备的表面活性剂制备水性环氧分散体
类似于分散体实例5实施分散体实例11,除了将9.5kg的实验1-型xz92585和0.5kg表面活性剂e-peg4600-585在夹套反应器中在95℃预共混。
所得
分散体实例12-使用在表面活性剂实例3中制备的表面活性剂制备水性环氧分散体
将9.7kg批量的der331和0.3kg表面活性剂e-peg4600-661在夹套反应器中在95℃预共混。一旦均匀,将材料冷却至80℃。然后将冷却的预共混物材料以60g/min泵送到转子定子混合器中。将e-sperse100阴离子表面活性剂(60%活性)流以1.75g/min泵送到混合器中。水也以10g/min添加到混合器中。
然后用额外的水稀释所得高内相乳液以获得60.41%的固体分散体。所获得
分散体实例13-使用在表面活性剂实例3中制备的表面活性剂制备水性环氧分散体
类似于分散体实例5实施分散体实例13,除了将9.7kg的den438和0.3kg表面活性剂e-peg4600-661在夹套反应器中在95℃预共混。
所得
分散体实例14-使用在表面活性剂实例3中制备的表面活性剂制备水性环氧分散体
类似于分散体实例5实施分散体实例14,除了将9.5kg的实验1-型xz92585和0.5kg表面活性剂e-peg4600-661在夹套反应器中在95℃预共混。
所得
分散体实例15-使用在表面活性剂实例4中制备的表面活性剂制备水性环氧分散体
将9.7kg批量的der331和0.3kg表面活性剂e-peg4600-664在夹套反应器中在95℃预共混。一旦均匀,将材料冷却至80℃。然后将冷却的预共混物材料以60g/min泵送到转子定子混合器中。将e-sperse100阴离子表面活性剂(60%活性)流以1.75g/min泵送到混合器中。水也以10g/min添加到混合器中。
然后用额外的水稀释所得高内相乳液以获得60.25wt%的固体分散体。所获得
分散体实例16-使用在表面活性剂实例4中制备的表面活性剂制备水性环氧分散体
类似于分散体实例5实施分散体实例16,除了将9.7kg的den438和0.3kg表面活性剂e-peg4600-664在夹套反应器中在95℃预共混。
所得
分散体实例17-使用在表面活性剂实例4中制备的表面活性剂制备水性环氧分散体
类似于分散体实例5实施分散体实例17,除了将9.5kg的实验1-型xz92585和0.5kg表面活性剂e-peg4600-664在夹套反应器中在95℃预共混。
所得
分散体实例18-使用在表面活性剂实例4中制备的表面活性剂制备水性环氧分散体
类似于分散体实例5实施分散体实例18,除了将der667(80wt%)、der330(5wt%)和propylcellosolv(15wt%)(可购自陶氏化学公司)的预共混物9.02kg与0.98kg表面活性剂e-peg4600-664在夹套反应器中在95℃预共混。
所得
表i描述并且图2示出了当使用具有不同mw的各种疏水物制造表面活性剂、该表面活性剂进而用于制造水性环氧分散体时,对水性环氧分散体的粘度性质的影响。
表i–水性环氧分散体
- 还没有人留言评论。精彩留言会获得点赞!