PARP抑制剂的结晶形式的制作方法
相关申请的交叉引用本申请要求2014年11月26日提交的美国临时申请no.62/084,652的权益,该临时申请的整个内容以引用方式并入本文。本公开涉及4,5,6,7-四氢-11-甲氧基-2-[(4-甲基-1-哌嗪基)甲基]-1h-环戊[a]吡咯并[3,4-c]咔唑-1,3(2h)-二酮及其盐的结晶形式。
背景技术:
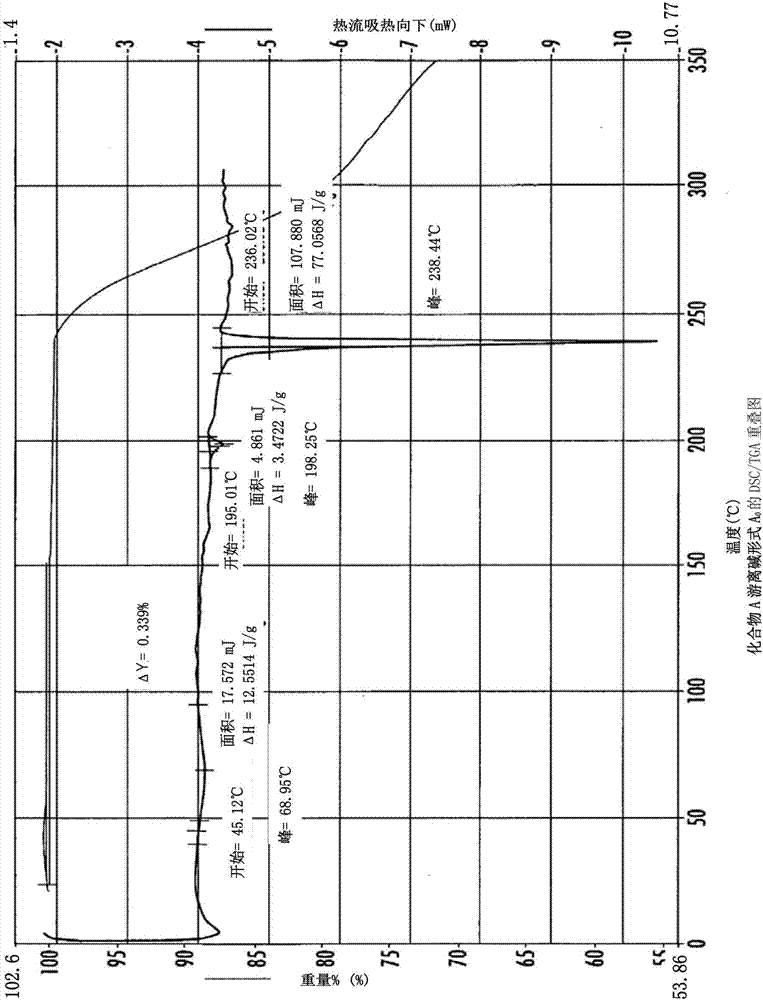
:化合物a(4,5,6,7-四氢-11-甲氧基-2-[(4-甲基-1-哌嗪基)甲基]-1h-环戊[a]吡咯并[3,4-c]咔唑-1,3(2h)-二酮)是用于单独地或联合化疗或放疗来治疗乳腺癌、卵巢癌和其他癌症的parp(多聚adp-核糖聚合酶)抑制剂。参见例如美国专利no.7,122,679、8,716,493和8,633,314。化合物a是化合物b的前药:化合物a的游离碱形式形成水合物,这是不可取的。此外,化合物a的游离碱形式具有低堆积密度,使得不便于制造。需要化合物a的替代形式。技术实现要素:本公开涉及化合物a乙酸盐形式a1.5、化合物a乙醇酸盐水合物形式a1、化合物al-苹果酸盐形式a1、化合物al-苹果酸盐形式a1.5、化合物al-焦谷氨酸盐形式a1、化合物a游离碱形式c0、化合物a盐酸盐形式a、化合物a富马酸盐形式a和化合物a对甲苯磺酸盐形式a。还描述了包含这些形式中的一种或多种的药物组合物。也描述了使用这些形式的方法。附图说明图1显示化合物a游离碱形式a0的xrpd图案。图2显示化合物a游离碱形式a0的dsc/tga重叠图。图3显示化合物a乙酸盐形式a1.5的xrpd图案。图4显示化合物a乙酸盐形式a1.5的vt-xrpd图案-请求模式(requestedmode)。图5显示化合物a乙酸盐形式a1.5的vt-xrpd图案-连续模式(continuousmode)。图6显示化合物a乙酸盐形式a1.5的dsc和tga重叠图。图7显示化合物a乙酸盐形式a1.5的dvs重叠图。图8显示化合物a乙酸盐形式a1.5的显微照片。图9显示化合物a乙醇酸盐水合物形式a1的xrpd图案。图10显示化合物a乙醇酸盐水合物形式a1的热xrpd图案。图11显示化合物a乙醇酸盐水合物形式a1的dsc和tga重叠图。图12显示化合物a乙醇酸盐水合物形式a1的dvs重叠图。图13显示化合物a乙醇酸盐水合物形式a1的显微照片。图14显示化合物al-苹果酸盐形式a1的xrpd图案。图15显示化合物a苹果酸盐形式a1的vt-xrpd图案。图16显示化合物al-苹果酸盐形式a1的dsc和tga重叠图。图17显示化合物al-苹果酸盐形式a1的dvs。图18显示化合物al-苹果酸盐形式a1的显微照片。图19显示化合物al-苹果酸盐形式a1.5的xrpd图案。图20显示化合物al-苹果酸盐形式a1.5的dsc和tga重叠图。图21显示化合物al-焦谷氨酸盐形式a1的xrpd图案。图22显示化合物al-焦谷氨酸盐形式a1的vt-xrpd图案。图23显示化合物al-焦谷氨酸盐形式a1的dsc和tga重叠图。图24显示化合物al-焦谷氨酸盐形式a1的dvs。图25显示化合物al-焦谷氨酸盐形式a1的显微照片。图26显示化合物a游离碱形式c0的xrpd图案。图27显示化合物a游离碱形式c0的热xrpd图案。图28显示化合物a游离碱形式c0的dsc和tga重叠图。图29显示化合物aa游离碱形式c0的显微照片。图30显示化合物a盐酸盐形式a的xrpd图案。图31显示化合物a盐酸盐形式a的dsc和tga重叠图。图32显示化合物a盐酸盐形式a的dvs。图33显示化合物a富马酸盐形式a的xrpd图案。图34显示化合物a富马酸盐形式a的dsc和tga重叠图。图35显示化合物a对甲苯磺酸盐形式a的xrpd图案。图36显示化合物a对甲苯磺酸盐形式a的dsc和tga重叠图。图37显示化合物b(1mg/kg静脉内)、化合物a抗坏血酸盐(30mg/kg口服)和化合物a乙醇酸水合盐(30mg/kg口服)在大鼠中的血浆水平。图38显示化合物a乙醇酸水合盐的单晶结构。具体实施方式本公开通过提供化合物a的新形式(包括化合物a的新结晶游离碱形式和化合物a的新结晶盐形式)而解决本领域中的需求。本公开尤其涉及化合物a乙酸盐形式a1.5、化合物a乙醇酸盐水合物形式a1、化合物al-苹果酸盐形式a1、化合物al-苹果酸盐形式a1.5、化合物al-焦谷氨酸盐形式a1、化合物a游离碱形式c0、化合物a盐酸盐形式a、化合物a富马酸盐形式a和化合物a对甲苯磺酸盐形式a。还描述了包含这些形式中的一种或多种的药物组合物。在一个实施方案中,本公开涉及化合物a乙酸盐形式a1.5。在一个方面,该结晶形式通过包括以下峰中的一个或多个的x射线衍射图案来表征:6.4、9.2、12.7、13.0、15.2、17.4、18.4、19.0、19.3、21.3、21.5、23.1、24.1、24.2和/或28.2±0.2度2-θ。在另一个方面,该结晶形式包括前述峰中的至少3个。在又一个方面,该结晶形式包括前述峰中的至少4、5、6、7、8、9或10个。在另一个方面,该结晶形式具有基本上如图3中所描绘的x射线粉末衍射图案。本公开还涉及化合物a乙醇酸水合盐。这些盐可在晶体结构中具有不同量的水。例如,化合物a与水的比率可为从约1:0.1至约1:1。在其他实施方案中,化合物a与水的比率为1:0.1、1:0.2、1:0.3、1:0.4、1:0.5、1:0.6、1:0.7、1:0.8、1:0.9或1:1。本公开的另一个实施方案涉及化合物a乙醇酸水合盐形式a1。在一个方面,该结晶形式通过包括以下峰中的一个或多个的x射线衍射图案来表征:8.1、8.2、8.7、13.9、14.7、14.9、16.3、17.4、17.6、18.2、18.5、19.0、20.2、20.6、21.2、21.4、23.0、24.5、24.7、26.1、26.3、28.0、30.0、30.1、30.2和/或32.8±0.2度2-θ。在另一个方面,该结晶形式包括前述峰中的至少3个。在又一个方面,该结晶形式包括前述峰中的至少4、5、6、7、8、9或10个。在另一个方面,该结晶形式具有基本上如图9中所描绘的x射线粉末衍射图案。本公开的又一个实施方案涉及化合物al-苹果酸盐形式a1。在一个方面,该结晶形式通过包括以下峰中的一个或多个的x射线衍射图案来表征:8.6、9.2、10.1、10.4、11.7、11.9、14.7、15.3、15.6、17.2、17.8、18.5、20.3、20.7、21.2、22.4、23.5、24.3和/或27.0±0.2度2-θ。在另一个方面,该结晶形式包括前述峰中的至少3个。在又一个方面,该结晶形式包括前述峰中的至少4、5、6、7、8、9或10个。在另一个方面,该结晶形式具有基本上如图14中所描绘的x射线粉末衍射图案。在另一个实施方案中,本公开涉及化合物al-苹果酸盐形式a1.5。在一个方面,该结晶形式通过包括以下峰中的一个或多个的x射线衍射图案来表征:5.5、6.8、8.0、8.4、8.8、9.2、11.8、12.8、13.1、13.6、14.4、16.0、16.7、18.1、18.5、19.4、20.2、20.5、21.1、21.9、23.4和/或24.6±0.2度2-θ。在另一个方面,该结晶形式包括前述峰中的至少3个。在又一个方面,该结晶形式包括前述峰中的至少4、5、6、7、8、9或10个。在另一个方面,该结晶形式具有基本上如图19中所描绘的x射线粉末衍射图案。本文还描述化合物al-焦谷氨酸盐形式a1。在一个方面,该结晶形式通过包括以下峰中的一个或多个的x射线衍射图案来表征:6.0、9.6、10.3、10.5、11.0、12.0、13.2、15.0、16.7、17.5、17.8、18.0、19.0、20.8、21.0、21.1、22.0、22.1、23.1、23.4、23.5、24.8和/或26.6±0.2度2-θ。在另一个方面,该结晶形式包括前述峰中的至少3个。在又一个方面,该结晶形式包括前述峰中的至少4、5、6、7、8、9或10个。在另一个方面,该结晶形式具有基本上如图21中所描绘的x射线粉末衍射图案。本公开还涉及化合物a游离碱形式c0。在一个方面,该结晶形式通过包括以下峰中的一个或多个的x射线衍射图案来表征:8.5、8.8、13.9、14.4、15.4、17.6、18.1、18.5、19.2、19.7、20.4、21.1、21.4、21.9、23.6、24.6、29.4和/或30.1±0.2度2-θ。在另一个方面,该结晶形式包括前述峰中的至少3个。在又一个方面,该结晶形式包括前述峰中的至少4、5、6、7、8、9或10个。在另一个方面,该结晶形式具有基本上如图27中所描绘的x射线粉末衍射图案。本公开的另一个实施方案涉及化合物a盐酸盐形式a。在一个方面,该结晶形式通过包括以下峰中的一个或多个的x射线衍射图案来表征:7.5、8.6、12.2、17.1、18.8、18.9、22.3、24.5、25.6、26.1、33.5和/或34.1±0.2度2-θ。在另一个方面,该结晶形式包括前述峰中的至少3个。在又一个方面,该结晶形式包括前述峰中的至少4、5、6、7、8、9或10个。在另一个方面,该结晶形式具有基本上如图30中所描绘的x射线粉末衍射图案。本公开的再一个实施方案涉及化合物a富马酸盐形式a。在一个方面,该结晶形式通过包括以下峰中的一个或多个的x射线衍射图案来表征:9.0、10.5、11.1、14.9、17.1、17.7、19.3、21.1、22.3、22.9、23.5、24.0、24.2、25.7、25.9、27.3、29.0和/或31.1±0.2度2-θ。在另一个方面,该结晶形式包括前述峰中的至少3个。在又一个方面,该结晶形式包括前述峰中的至少4、5、6、7、8、9或10个。在另一个方面,该结晶形式具有基本上如图33中所描绘的x射线粉末衍射图案。本公开的还一个实施方案涉及化合物a对甲苯磺酸盐形式a。在一个方面,该结晶形式通过包括以下峰中的一个或多个的x射线衍射图案来表征:6.0、9.6、10.3、10.5、11.0、12.0、12.9、13.2、15.0、16.7、17.0、17.5、17.8、18.0、19.0、20.8、21.0、21.1、22.1、22.7、23.1、23.4、23.5、24.8和/或26.6±0.2度2-θ。在另一个方面,该结晶形式包括前述峰中的至少3个。在又一个方面,该结晶形式包括前述峰中的至少4、5、6、7、8、9或10个。在另一个方面,该结晶形式具有基本上如图35中所描绘的x射线粉末衍射图案。在一些实施方案中,本公开的多晶型形式基本上不含任何其他多晶型形式或指定的多晶型形式。在本发明的任何实施方案中,所谓“基本上不含”是指本发明的所述形式含有20%(w/w)或以下、10%(w/w)或以下、5%(w/w)或以下、2%(w/w)或以下、尤其是1%(w/w)或以下、更尤其是0.5%(w/w)或以下且最尤其是0.2%(w/w)或以下的任何其他多晶型物或指定的一种或多种多晶型物。在其他实施方案中,本公开的多晶型物含有从1%至20%(w/w)、从5%至20%(w/w)或从5%至10%(w/w)的任何其他多晶型物或指定的一种或多种多晶型物。本发明的盐和固态形式具有有利的性质,包括以下至少一种:高结晶度、溶解度、溶解速率、形态、针对多晶型转化和/或针对脱水的热和机械稳定性、储存稳定性、低残余溶剂含量、较低的吸湿度、流动性以及有利的加工和处理特性,诸如可压缩性和堆积密度。晶型在本文中可称为通过“基本上如图中所描绘的”图形数据来表征。这些数据包括例如粉末x射线衍射图。技术人员将理解的是,数据的此类图形表示可以发生小的变化,例如在峰相对强度和峰位置上,原因是诸如仪器响应中的变化和样品浓度及纯度中的变化等因素,这对于技术人员而言是熟知的。尽管如此,技术人员将能够容易地比较本文图中的图形数据与针对未知晶型产生的图形数据并确认两组图形数据是表征相同的晶型还是两种不同的晶型。如本文所用的术语“无定形的”是指缺乏特征性晶体形状或结晶结构。如本文所用的术语“结晶的”是指具有规则性重复的分子排列或外晶面平面(externalfaceplane)。如本文所用的术语“结晶形式”是指一种固体化合物或多种化合物的混合物,当通过x射线粉末衍射对其分析时,这种物质提供特征性峰图案;这种物质包括但不局限于多晶型物、溶剂化物、水合物、共晶和去溶剂化的溶剂化物。术语“多晶型的”或“多晶型现象”被定义为对于相同的化学分子有至少两种不同的结晶排列的可能性。如本文所用的术语“溶液”是指含有至少一种溶剂以及至少一种化合物的混合物,该化合物在该溶剂中至少部分地溶解。如本文所用的术语“药学上可接受的赋形剂”包括任何及所有的溶剂、分散介质、包衣、抗细菌剂和抗真菌剂、等渗剂和吸收延迟剂等。对于药物活性物质而言,此类介质和药剂的使用在本领域中是熟知的,诸如在remington:thescienceandpracticeofpharmacy,第20版;gennaro,a.r.编,lippincottwilliams&wilkins:philadelphia,pa.,2000中。除非在任何常规介质或药剂与活性成分不相容的情况下,否则考虑了它在治疗性组合物中的使用。还可以将补充性活性成分掺入这些组合物中。本发明的药物组合物可以多种方式使用,包括但不限于增强辐照或dna损伤性化疗剂的抗肿瘤活性(griffin,r.j.;curtin,n.j.;newell,d.r.;golding,b.t.;durkacz.b.w.;calvert,a.h.theroleofinhibitorsofpoly(adp-ribose)polymeraseasresistance-modifyingagentsincancertherapy.biochemie1995,77,408)。出于治疗性目的,本发明的结晶形式可以通过导致活性剂与受试者体内的药剂作用位点相接触的任何手段来施用。结晶形式可以通过可用于与药物结合使用的任何常规手段作为单独的治疗剂或与其他治疗剂比如止痛剂联合来施用。本发明的结晶形式优选地以用于治疗本文所述的疾病和病症的治疗有效量施用给对其有需要的受试者。在治疗性或预防性用途中,本发明的结晶形式可以通过通常施用药物的任何途径来施用。此类施用途径包括腹膜内、静脉内、肌内、皮下、鞘内、气管内、心室内、口、颊、直肠、肠胃外、鼻内、透皮或真皮内。施用可以是全身或局部的。本文所述的结晶形式可以采取纯的形式、与其他活性成分组合、或与药学上可接受的无毒赋形剂或载体组合施用。口服组合物一般将包含惰性稀释载体或可食用载体。可以包含药学上相容的粘合剂和/或佐剂材料作为组合物的一部分。片剂、丸剂、胶囊、锭剂等可以含有任何下述成分或性质类似的化合物:粘合剂,诸如微晶纤维素、黄蓍胶或明胶;赋形剂,诸如淀粉或乳糖;分散剂,诸如藻酸、primogel或玉米淀粉;润滑剂,诸如硬脂酸镁;助流剂,诸如胶体二氧化硅;甜味剂,诸如蔗糖或糖精;或调味剂,诸如胡椒薄荷、水杨酸甲酯或橙香精。当剂量单位形式是胶囊时,除了上述类型的材料之外,它还可以含有液体载体,诸如脂肪油。此外,剂量单位形式可以含有改变剂量单位的物理形式的各种其他材料,例如糖、虫胶或肠溶试剂的包衣。此外,除了活性化合物之外,糖浆还可以含有作为甜味剂的蔗糖以及某些防腐剂、色素、着色剂和调味剂。用于施用的备选制剂包括无菌水性或非水性溶液、悬浮液和乳液。非水性溶剂的实例是二甲基亚砜、醇类、丙二醇、聚乙二醇、植物油诸如橄榄油和注射用有机酯诸如油酸乙酯。水性载体包括醇类与水的混合物、缓冲介质和盐水。静脉内溶媒包括流体和营养补充剂、电解质补充剂,诸如基于林格氏(ringer's)葡萄糖等的那些。也可以存在防腐剂和其他添加剂,比如抗微生物剂、抗氧化剂、螯合剂、惰性气体等。结晶形式向哺乳动物施用的优选方法包括腹膜内注射、肌内注射和静脉内输注。各种液体制剂可能用于这些递送方法,包括盐水、醇、dmso和水基溶液。浓度可以根据待递送的剂量和体积而变化,并且可以在从约1至约1000mg/ml的范围内。液体制剂的其他成分可以包括防腐剂、无机盐、酸、碱、缓冲剂、营养剂、维生素或其他药物诸如止痛剂或另外的parp和激酶抑制剂。因此,已经参照了特定的优选实施方案和示例性实例描述了本发明,但是本领域的技术人员可以认识到不脱离如说明书中所公开的本发明精神和范围的对所描述和例示的发明的适当修改。示出实施例以有助于理解本发明,但无意且不应被视为以任何方式限制本发明的范围。实施例以下实施例中所用的溶剂具有试剂级质量且无需进一步纯化即使用。化合物a的已知形式用a0和b0来表示无水材料,用hd来表示水合物。x-射线粉末衍射(xrpd)标准反射模式测量:在45kv和40ma下使用cukα辐射在配有x'celerator检测器的panalyticalxpertpro衍射仪上记录粉末x射线衍射图案。kα1辐射使用高度取向的晶体(ge111)入射光束单色仪获得。将10mm光束掩模以及固定的(1/4°)发散和防散射(1/8°)狭缝插到入射光束一侧上。将固定的5mm接收狭缝和0.04弧度soller块插到衍射光束一侧上。x射线粉末图案扫描从约2至40°2θ以0.0080°的步长和96.06秒的计数时间(这导致约0.5°/min的扫描速率)来收集。在硅零背景(zbg)板上展开样品以用于测量。用panalyticalpw3064旋转器(15转/分钟)旋转样品。在收集数据之前对si参比标准进行的测量得到了2θ以及强度的值,这些值完全处于28.44<2θ<28.50的容限内并且显著大于150cps的最小峰高。scxrd-单晶x射线衍射:对于数据收集,从由约三或四个单独块构成的团块脱落下一块(0.12×0.04×0.03mm3)以得到看起来为单晶的晶体。借助聚异丁烯油(也称为paratone)将晶体安装到bruker-noniusx8proteum衍射仪上的细玻璃纤维上,衍射仪附连到noniusfr-591旋转阳极(cuka),该阳极具有‘helios’聚焦光学器件。将晶体通过得自cryoindustriesofamerica的cryocoollt2自始至终维持在90k。用于索引的衍射图像清楚地显示出分裂反射(splitreflection),与开裂(cracking)或孪晶(twinning)相符,但是具有靠得足够近而融合在一起的光斑分量(spotcomponent)。数对分裂反射的相对强度表明开裂比孪晶的可能性更大。从存在于72个衍射图像(六组,每组12个0.5°的帧)中的反射来索引晶体。数据采集包括以三个检测器偏转角在15次扫描中获得的1485个2°的帧(以2θ在-40°进行的两个扫描,以2θ在-45°进行的三个90°ω-扫描,以2θ在-96°进行的四个扫描,以及以2θ在-96°进行的六个90°ω-扫描),其足够以四倍冗余度覆盖任意取向的三斜晶体的倒易空间,达到的分辨率。使用得自bruker-axs的apex2包中的程序,对数据进行积分、换算、平均化和合并。最终的晶胞参数来源于积分过程的输出诊断。结构通过标准直接方法使用shelxs进行解析,并使用shelxl进行精修,两者均得自shelx97包。使用得自shelxtl套装的xp和得自ccdc的mercury绘图。通过platon进行了另外的分子图形和空间计算。对所有非氢原子的位置和各向异性位移参数进行了精修。h原子位于差分傅里叶图谱(differencefourier’smap)中,但附连到碳原子的那些氢原子在几何上重新定位。h原子最初通过软约束(softrestraint)在键长和角度上进行精修,以调整它们的几何(n-h和0.93-0.98范围内的c-h至和uiso(h)(在1.2-1.5倍母原子ueq的范围内),之后,通过跨骑约束(ridingconstraint)对位置进行精修。单晶单元晶胞参数针对测得的xprd图案进行默认reitveld精修得到了良好的拟合,没有无法解释的峰。变温x射线粉末衍射(vt-xrpd):通过antonpaarchc温度/湿度箱在通过antonpaartcu110温度控制单元进行的计算机控制下执行变温研究。一般来讲,在氮气流过照相机的情况下进行测量。使用了两个测量方案:受限和连续。在受限模式中,仅在chc箱达到请求温度后才进行测量。在连续模式中,将样品以10℃/分钟加热,并随着温度的变化进行快速扫描。在两种情况下,在达到请求的温度后,将样品以35℃/分钟冷却,并在25℃进行缓慢扫描。从约3至30°或40°采集缓慢2θ扫描,步长为0.0080°,计数时间为100.97秒,这导致0.5°/min的扫描速率。从约3至30°2θ采集快速扫描,步长为0.0167°,计数时间为1.905秒,这导致约44°/min的扫描速率。所选的温度基于dsc结果。对于衍射仪设置,将10mm光束掩模、0.04弧度soller狭缝以及固定的(1/4°)发散和防散射(1/8°)狭缝插到入射光束一侧上。将固定的5mm接收狭缝、0.04弧度的soller狭缝和0.02mm的镍滤光片插到衍射光束一侧上。差示扫描热量法(dsc):热曲线使用在分析之前用铟校准的运行pyris软件6.0版的perkin-elmersapphiredsc单元获得,该dsc单元配有自动进样器。称取1-10mg的固体样品加到20μl的铝质针孔样品盘中。然后将dsc单元用氮气吹扫并且将温度以10℃/min从0℃加热到270℃。将铟(tm=156.6℃,δhfus=28.45jg-1)用于校准。调制式差示扫描量热法(mdsc):使用taq200调制dsc单元采集热曲线。称取5-20mg固体样品加到50μl铝针孔气密封盘中。然后将mdsc单元用氮气吹扫,并将温度以2℃/min从0℃加热至350℃,加热速率为2℃/min,在60秒周期内的调制幅度为+/-1℃。热重量质谱法(tga/ms):使用由亚铝美(alumel)(95%镍、2%锰、2%铝和1%硅)、镍和草酸钙一水合物校准的运行pyris软件6.0版的perkin-elmerpyris1tga单元来获得热曲线。随着在用氦气以约50ml/min吹扫的炉中以10℃/min从25℃加热至250℃,对1-5mg之间的tga样品进行百分比重量损失的监测。为了同时跟踪在所研究的温度范围内气体分解产物的释放,将热天平连接到thermostar四极杆质谱仪(asslar,germany)。将气态分解产物引入到质谱仪中的传输管线是温度控制到200℃以避免可能的所释放气体的冷凝的去活化熔融石英毛细管(sgeanalyticalscience,熔融石英(100%甲基去活化),220mmod,150mmid,australia)。以此方式,可以同时记录所选离子种类的质谱离子强度曲线和tga重量损失。动态蒸汽吸附(dvs):使用dvs-ht仪器(surfacemeasurementsystems,london,uk)进行了gvs实验。该仪器使用质量分辨率为±0.1μg的记录超微量天平来在重量上测量蒸汽的吸收和损失。通过使用电子质量流量控制器混合饱和的干燥载气流来控制样品周围的蒸汽分压(±1.0%)。将期望的温度保持在±0.1℃。在该期望的温度下,将样品(1至10mg)置于dvs-ht和dvs-1仪器中。在40%rh和25℃下(典型的室内条件)加载和卸载样品。如下所列(给出1个完整循环的2次扫描)进行水分吸着等温线绘制。软件使用最小二乘法最小化程序以及质量松弛模型来预测渐进值。测得的质量平衡值在转到下一个%rh值之前必须在由软件预测的值的2%之内。将最小平衡时间设为1小时,最大平衡时间设为4小时。光学显微术:使用olympusb60偏光显微镜来进行样品形态的显微观察。将样品悬于矿物油中并在观察之前用盖玻片压在载玻片上。使用fw-24(paxcam)照相机来采集图像。10x物镜加上来自显微镜光学器件的另外10x放大给出100x的总放大。使用pax-it软件(6.2版)来捕获并分析图像。核磁共振光谱法(1h-nmr):通过1h-nmr光谱法测定盐的化学计量,其中使用在针对每个样品进行了优化以得到可能最佳的光谱的条件下运行的brukerdpx400仪器。将每个样品(2-4mg)溶于0.75mldmso-d6,并在薄壁玻璃管(4×14mm)中获得光谱。通过hplc进行鉴定、分析和纯度测定设备:测试在经过校准和验证的名为lc-0430-ad或lc-418-1d的agilent1200快速分离高效液相色谱(hplc)系统上进行。系统包括二元sl泵、脱气器、具有级分收集器的高效自动进样器sl、具有双阀柱切换器的恒温柱温箱和dadsl检测器。所有标准溶液和样品均在a类玻璃容量瓶中配制并置于自动进样器小瓶中。使用经校准的mettler分析天平进行标准称量。使用eppendorf微量离心机对制备样进行离心。采集原始色谱数据,并使用empower2软件积分。将microsoftofficeexcel2003用于计算结果。试剂:乙腈得自cci。三氟乙酸得自emd。hplc级水(18mω·cm)得自位于实验室a211的实验室barnsteadnanopure系统upw-0403-ad。如之前所述制备化合物a和b。仪器参数:盐在40℃和75%湿度下的固态稳定性:称取待研究形式的样品(15-20mg)加到标准1.5mlhplc小瓶(32×11.6mm)中,不加盖在40℃和75%rh的稳定箱中储存0、7、14和28天。在指定的天数取出样品并盖上。对每个时间点的样品通过纯度和分析测量法完成了xrpd、dsc、tga和hplc鉴定测量。水溶性估计:称取多份十毫克待研究的盐形式加到标准1.5mlhplc小瓶(32×11.6mm)中。将搅拌棒和100μl水加到每只小瓶中。将样品盖上并搅拌5-10分钟。如果目测未得到透明溶液,则添加另外100-300μl部分的水并搅拌。重复该过程,直到样品溶解或直到添加了1000μl水。基于溶解已知重量的样品所必需的水的体积来估计溶解性。得自这些测量的结果在表11中给出。表1.通过缓慢冷却在丙酮中形成的具有一当量酸的盐的估计水溶性和hplc分析实施例1.通过成熟在丙酮中形成具有两当量酸的盐在五只20ml闪烁小瓶中的每一只的15ml丙酮中,将200mg化合物a(0.478毫摩尔)通过温热和搅拌而溶解。将1.95当量的乙酸、乙醇酸、l-苹果酸或l苹果酸(1当量,0.48毫摩尔)加到透明的化合物a溶液中。添加完这些酸后,透明溶液就变浑浊且开始结晶。使小瓶在hel单元上接受两个成熟循环。每个成熟循环包括在一小时的时间段内加热到50℃,在50℃保持四小时,在一小时的时间段内冷却到5℃,并在5℃保持四小时。通过抽吸过滤而分离固体,将固体在50℃和清扫真空(housevacuum)(~200mm)下干燥过夜得到黄色固体。结果在表2中给出。表2样品酸xrpddsc,℃tga,%估计的水溶性39-1(2)乙酸a1.5185.224.4~25mg/ml39-2(2)乙醇酸a168.9,205.44.8>100mg/ml39-3(2)l-苹果酸a1186.43.6>100mg/ml39-5(2)l-苹果酸(1当量)a1+c0186.51.0>100mg/ml实施例2.使用快速冷却在丙酮中进行酸筛选(两当量)向七只容纳有搅拌棒和1.5ml化合物a溶液(13.3mg/ml)的hplc小瓶中,称取将得到两当量(0.096毫摩尔)的酸量,或通过移液器添加。将样品盖上并加热到沸点,然后在2-8℃的冰箱中冷却过夜。通过抽吸过滤而分离固体,将固体在50℃和清扫真空(~200mm)下干燥过夜得到黄色固体。结果在表3中给出。表3实施例3.通过浆液转化在丙酮中形成具有两当量酸的盐在五只具有18ml丙酮的20ml玻璃闪烁小瓶的每一只中,将400mg化合物a(0.956毫摩尔)温热并搅拌而浆液化。将两当量的乙酸、乙醇酸、l-苹果酸、l-焦谷氨酸或l-苹果酸(1当量,0.956毫摩尔)加到每只小瓶中的化合物a悬浮液中。将这些混合物盖上并温热至接近沸点。在所有情况下,均注意到厚重的黄色固体。使样品在试验台上冷却至环境温度,然后在2-8℃的冰箱中冷却过夜。通过抽吸过滤而分离固体,将产物在50℃和清扫真空(~200mm)下干燥过夜得到黄色固体。结果在表4中给出。表4样品酸xrpddsc,℃tga,%估计的水溶性39-1乙酸a1.5185.4,分裂峰2.1~50mg/ml39-2乙醇酸a177.4,209.01.9<10mg/ml39-3l-苹果酸a1193.33.6>100mg/ml39-4l-焦谷氨酸a150.4,198.23.5>100mg/ml39-5l-苹果酸(1当量)a1+c0192.21.0>100mg/ml实施例4.在丙酮中进行酸筛选(两当量)-成熟通过在磁力搅拌棒搅拌下温热,在18ml丙酮中溶解240mg化合物a(0.574毫摩尔)。将该溶液等量分配到12只1.5mlhplc小瓶中。向5只容纳有化合物a溶液的等分试样和搅拌棒的小瓶的每一只中,称取将得到两当量(0.096毫摩尔)的酸量,或通过移液器添加。将样品盖上,并使样品在hel单元上接受两个成熟循环。每个成熟循环包括在一小时的时间段内加热到50℃,在50℃保持四小时,在一小时的时间段内冷却到5℃,并在5℃保持四小时。通过抽吸过滤而分离固体,将固体在50℃和清扫真空(~200mm)下干燥过夜得到黄色固体。结果在表5中给出。表5样品酸xrpddsc,℃tga,%估计的水溶性30-1乙酸a1,5187.7,334.121.7~20mg/ml30-2乙醇酸a1206.63.2>100mg/ml30-3l-苹果酸a1190.21.5>100mg/ml30-4l-焦谷氨酸a1197.51.8>100mg/ml30-5l-苹果酸(1当量)c0207.32.2~25mg/ml实施例5.丙酮中一当量-缓慢冷却在12ml丙酮中配制240mg化合物a(0.57毫摩尔)的溶液,并在搅拌下温热以溶解。十二个该溶液的均等等分试样将在每只小瓶中的1ml丙酮中将得到20mg(0.0478毫摩尔)化合物a。称取对应于1.05当量(0.06毫摩尔)酸的酸重量加到12只1.5mlhplc小瓶中,或如果为液体则通过移液器添加。向每只小瓶中,添加化合物a的一个等分式样。将小瓶盖上并在搅拌下温热以混合,然后在hel单元上接受2个缓慢冷却循环。hel单元上的每个缓慢冷却循环包括:在1小时的时间段内加热到80℃,在80℃下保持1小时,然后在5小时的时间段内冷却到5℃,并在5℃下保持16-18小时。通过抽吸过滤而分离固体,将样品在50℃和清扫真空(~200mm)下干燥过夜。结果在表6中给出。表6*exo=放热实施例6.制备抗坏血酸盐称取200mg化合物a(0.478毫摩尔)在搅拌棒搅拌下加到20ml玻璃闪烁小瓶中,然后添加88.4mg(0.503毫摩尔,1.05当量)抗坏血酸(j.t.baker,无水,批号b36597)。通过移液器添加2.5ml2,2,2-三氟乙醇,并将样品温热。使所形成的浆液在hel单元上接受2个缓慢冷却循环。hel单元上的每个缓慢冷却循环包括:在1小时的时间段内加热到80℃,在80℃下保持1小时,然后在5小时的时间段内冷却到5℃,并在5℃下保持16-18小时。通过抽吸过滤而分离固体,将样品在50℃和清扫真空(~200mm)下干燥过夜得到142mg黄色固体(49%收率)。通过hplc分析结晶产物,得到96.2%的化合物b和0.8%的化合物a。化合物b盐的结构通过1h-nmr确认。化合物a游离碱形式a0xrpdxrpd在图1中描绘。热分析热数据在图2中描绘。化合物a乙酸盐形式a1.5制备根据实施例1中的程序制备盐。xrpd乙酸盐形式a1.5的x射线衍射数据在图3和表7中给出。以请求模式(165℃和200℃)进行的变温xrpd测量显示出形式上的两个变化-从乙酸盐到形式b0,然后转化成形式a0。在连续模式中,使用从5.5°至11.5°的一分钟扫描和1℃/分钟温度斜坡,注意到形式上的三个变化:乙酸盐到游离碱b0、b0到a0以及a0到无定形(图4)。乙酸盐在从91℃至130℃的温度范围内缓慢转化成游离碱形式b0。形式在197℃与200℃之间从b0变成a0(图5)。表7.乙酸盐形式a1.5的xrpd峰*使用zbg或玻璃板通常引入正样品高度位移并导致2θ值的小(0.05°至0.2°)偏移。最高的峰(强度100%)以黑体字母示出。热分析乙酸盐形式a1.5的dsc曲线显示出在185.4℃处存在一个吸热/降解峰,δhfus为172.0j/g(图6)。乙酸盐在25与150℃之间具有29.5%的重量损失。吸水图7中的dvs图线表明样品似乎从开始就饱和。在干燥曲线中存在稳定的重量损失,未达到平衡。对于每个循环,将样品在0%rh下干燥4小时。运行了4个循环,显示出连续的重量损失。在另一dvs单元上重复实验,显示出类似的结果。1h-nmr光谱1h-nmr光谱显示出化合物a预期的所有峰。将在约7.5ppm处的峰归一化到预期在该区域中吸收的一个芳族质子。与化合物相关的其余峰然后遵循正确的比率。对于乙酸盐,预期仅有一个在1.9-2.0ppm处的峰。该峰应该集成了3个质子。相反,其显示出约4.5个质子,每个化合物a分子约1.5个乙酸分子。稳定性表8中给出了乙酸盐形式a1.5在40℃和75%rh下老化的数据。xrpd在整个28天的测试期中发生变化。tga和化合物a分析值可能反映乙酸的损失,如在上述热和xrpd研究中所见。dsc、hplc纯度和化合物b分析在研究中相对恒定。单乙酸盐应分析为87.5%的化合物a。二乙酸盐应分析为77.7%的化合物a。表8中的值表明盐随着老化而改变组成。1h-nmr测得每分子化合物a具有1.5分子乙酸。xrpd图案显示出水合物化合物a游离碱形式hd的峰。可能的是,随着样品老化,过量的乙酸挥发。乙酸的挥发性和变化的xrpd图案表明要选择另一种候选物。表8.乙酸盐形式a1.5在40℃和75%rh下的稳定性光学显微术如图8中所示的样品展示出不规则形状的晶体的团聚体。样品在平面偏振光下显示出双折射。化合物a乙醇酸盐水合物形式a1制备根据实施例1制备盐。xrpd乙醇酸水合盐形式a1的x射线衍射数据在图9和表9中给出。变温xrpd测量的重叠图扫描在图10中示出。初始xrpd图案与乙醇酸盐水合物形式a1进行比较。在暴露于干燥n2气氛后无变化。在175℃下的一个小时缓慢扫描测量期间,图案发生变化。在从175℃加热至225℃后,峰强度增加。未与已知的化合物a游离碱图案进行比较。在测量结束时板上的样品为深棕色粉末,其并无发生熔化的外观。在加热到175℃和225℃后观察到的图案与化合物b进行部分比较。这与显示出在130℃后变化并在205℃时熔化的dsc相符。vt-xrpd和dsc均与乙醇酸损失且转化成化合物b相符。表9.乙醇酸水合盐形式a1的xrpd峰*使用zbg或玻璃板通常引入正样品高度位移并导致2θ值的小(0.05°至0.2°)偏移。最高的峰(强度100%)以黑体字母示出。单晶结构单晶x射线结构确认了存在乙醇酸根阴离子,并表明哌嗪氮原子携带氢原子。分子在图38中示出。结构还显示以60%的占比存在的水分子,也就是说,化合物a与水的比率为1:0.6。结构细节在下表中给出。化合物a乙醇酸盐水合物的非氢原子的分数坐标和各向同性位移参数见下。化合物a乙醇酸盐水合物的氢原子的分数坐标和各向同性位移参数见下。热分析乙醇酸水合盐形式a1的dsc曲线显示存在两个不同的吸热峰:一个在77.4℃处,δhfus为63.4j/g;第二个峰在209.0℃处,δhfus为170.9j/g(图11)。乙醇酸水合盐在25与150℃之间具有1.9%的重量损失。吸水图12中的dvs图线表明在整个rh范围内存在表面吸附,而体吸收有限。在90%rh下总的水分吸收为~3.5%。1h-nmr光谱光谱给出化合物a所有必要的峰。在将积分归一化到化合物a在约7.5ppm处的芳族区中的一个质子后,在约3.9ppm处存在一个双质子单重峰,其对应于与乙醇酸的亚甲基基团相关的两个质子。这表明盐中化合物a与乙醇酸的摩尔比为1:1。稳定性表10中给出的数据表明该盐对测试条件相当稳定。在28天后注意到化合物b中的适度增加。如1h-nmr所表明,单乙醇酸盐应具有84.5%化合物a的化合物a分析结果。tga中增加的损失表明含水量的增加,例如,对于1:1的水与化合物a比率,预期将有3.5%的损失。表10.乙醇酸盐水合物形式a1在40℃和75%rh下的稳定性光学显微术在图13中,样品展示出单独的晶体和晶体的团聚体。样品在平面偏振光下显示出双折射。化合物al-苹果酸盐形式a1制备根据实施例1制备盐。xrpd苹果酸盐形式a1的x射线衍射数据在图14和表11中给出。vt-xrpd研究的重叠图缓慢扫描在图15中示出。初始xrpd图案如预期一样。在暴露于干燥n2气氛后形式上无变化(图15)。当将样品在175℃下保持一小时时,存在变化。在首次达到175℃时进行的快速扫描与开始图案进行比较。在175℃后进行的快速扫描中,结晶几乎完全消失。在加热到175℃并冷却到25℃后该样品所观察到的缓慢扫描图案与化合物b的图案进行部分比较。该观察结果与化合物b的热分解相符。表11.苹果酸盐形式a1的xrpd峰*使用zbg或玻璃板通常引入正样品高度位移并导致2θ值的小(0.05°至0.2°)偏移。最高的峰(强度100%)以黑体字母示出。热分析苹果酸盐形式a1的dsc曲线显示出在186.4℃处存在一个吸热峰,δhfus为75.7j/g(图16)。马来酸盐在25与150℃之间具有1.0%的重量损失。吸水图17中的dvs图线表明在从40%rh至70%rh的第一轮循环中存在极少的吸水。仅发生表面吸附。在80%rh下,吸水增加。由于体吸收存在大的滞后间隙(hysteresisgap)。总吸收为~2%。等温线不可逆。1h-nmr光谱存在化合物a预期的所有峰。在7.5ppm处的一个芳族质子的归一化后,在约4.05ppm处存在一个单质子三重峰,与l-苹果酸一致。这确立了呈形式a1的化合物al-苹果酸盐的1:1化学计量。稳定性表12中的数据显示,l-苹果酸盐对测试条件稳定,具有恒定的xrpd、dsc、tga和hplc纯度值(mjj3331-49)。在28天后观察到化合物b中的增加。与乙醇酸水合盐一样,化合物a的l-苹果酸盐分析值低于75.8%的预期值。表12.l-苹果酸盐形式a1在40℃和75%rh下的稳定性光学显微术在图18中,样品显示出单独的晶体和不规则形状的晶体的团聚体。样品在平面偏振光下显示出双折射。化合物al-苹果酸盐形式a1.5制备根据实施例2制备盐。xrpd苹果酸盐形式a1.5的x射线衍射数据在图19和表13中给出。表13.苹果酸盐形式a1.5的xrpd峰*使用zbg或玻璃板通常引入正样品高度位移并导致2θ值的小(0.05°至0.2°)偏移。最高的峰(强度100%)以黑体字母示出。热分析l-苹果酸盐形式a1.5的dsc曲线显示出在160.4℃处存在一个吸热峰,δhfus为39.2j/g(图20)。l-苹果酸盐在25与150℃之间具有3.6%的重量损失。该形式在低得多的温度下熔化并具有比苹果酸盐形式a1更大的重量损失。1h-nmr光谱l-苹果酸盐形式a1.5的1h-nmr光谱表明存在化合物a的所有峰,并且归一化积分表明两分子的化合物a存在约3分子的l-苹果酸。该制备代表了化合物al-苹果酸盐的新形式。化合物al-焦谷氨酸盐形式a1制备根据实施例3制备盐。xrpdl-焦谷氨酸盐形式a1的x射线衍射数据在表14和图21中给出。xrpd图案表明高度结晶的固体。变温xrpd测量结果在图22中示出。初始xrpd图案如预期一样。在加热到175℃后形式上无变化。在实验结束时,将黑色玻璃留在zbg板上。对化合物b的预期图案与加热到210℃后的样品进行比较显示出小的差异。这表明化合物a转化成了化合物b和可能的第二组分。表14.l-焦谷氨酸盐形式a1的xrpd峰*使用zbg或玻璃板通常引入正样品高度位移并导致2θ值的小(0.05°至0.2°)偏移。最高的峰(强度100%)以黑体字母示出。热分析l-焦谷氨酸盐形式a1的dsc曲线显示存在两个吸热峰:一个在50.4℃处,δhfus为35.6j/g,一个在198.2℃处,δhfu为76.8j/g(图23)。焦谷氨酸盐在25与150℃之间具有3.5%的重量损失。吸水在dvs图线(图24)中,表明在第一个循环中,在40-75%的rh范围内存在极少的吸水(~2%)。仅发生表面吸附。在80%rh下,存在大量的吸水。在50-90%rh下的大的滞后间隙是由于体吸收而可能形成了水合物。总吸收为~27%。1h-nmr光谱存在化合物a的所有峰。在将积分归一化到化合物a在约7.5ppm处的芳族峰中的一个质子后,在约7.85ppm处存在另外一个单质子单重峰,其对应于焦谷氨酸中的酰胺氮上的氢原子。此外,在约4.05ppm处存在另外一个单质子多重峰,其来自附连到与羧酸基团相邻的碳原子上的一个氢原子。这确立了该盐为化合物a的单l-焦谷氨酸盐。稳定性该盐在28天的测试期中稳定,但化合物b含量缓慢增加(表15)。表15.l-焦谷氨酸盐形式a1(用两当量的酸制备)在40℃和75%rh下的稳定性光学显微术如图25中所示,样品展示出不规则形状的晶体的团聚体。样品在平面偏振光下显示出双折射。盐的比较在表16中,比较了乙醇酸盐水合物形式a1、l-苹果酸盐形式a1以及l-焦谷氨酸盐形式a1的一当量和两当量制备物。乙醇酸水合盐形式a1在40℃和75%rh稳定性测试期间生成了最少量的化合物b。乙醇酸盐水合物表现出偏向于吸水,因为在稳定性测试期间tga值增至3.5%(表10)。表16.化合物a盐的比较化合物a游离碱形式c0制备根据实例4制备游离碱。xrpd游离碱形式c0的x射线衍射数据在图26和表17中给出。xrpd图案显示出结晶固体。变温xrpd测量结果在图27中示出。初始xrpd图案与形式c0的预期图案进行比较。在暴露于干燥n2气氛后形式上无变化。在加热到175℃后形式上无变化。在加热到235℃后,xrpd图案发生变化并类似于但不同于化合物b观察到的图案。对于其他vt样品,看见了相似的图案。似乎在在该分解产物中存在两种组分。表17.游离碱形式c0的xrpd峰热分析游离碱形式c0的dsc曲线显示出在207.3℃处存在一个吸热峰,δhfus为71.4j/g(图28)。形式c0在25与150℃之间具有2.3%的重量损失。光学显微术在图29中,样品展示出团聚体和单独的不规则形状的晶体。样品在平面偏振光下显示出双折射。化合物a盐酸盐形式a制备根据实施例5制备盐。xrpd氯化盐形式a的x射线衍射数据在图30和表18中给出。表18.盐酸盐形式a的xrpd峰*使用zbg或玻璃板通常引入正样品高度位移并导致2θ值的小(0.05°至0.2°)偏移。最高的峰(强度100%)以黑体字母示出。热分析盐酸盐形式a的dsc曲线显示出在247.3℃处存在一个吸热峰,δhfus为41.6j/g(图31)。盐酸盐形式a在25与150℃之间具有0.2%的重量损失。吸水dvs图线(图32)表明在整个rh范围内存在表面吸附,而体吸收有限。总的水分吸收为~2.25%。稳定性表19中的数据显示相对恒定的xrpd图案和dsc值,tga值适度变化。hplc值与分析值大不相同,在28天测试后降至近一半。还注意到hplc纯度的平稳下降,且化合物b含量增至1.5%。化合物a单盐酸盐中的化合物a含量的理论值为92.0%。表19.盐酸盐形式a在40℃和75%rh下的稳定性化合物a富马酸盐形式a制备根据实施例5制备盐。xrpd化合物a富马酸盐形式a的x射线衍射数据在图33和表20中给出。表20.富马酸盐形式a的xrpd峰*使用zbg或玻璃板通常引入正样品高度位移并导致2θ值的小(0.05°至0.2°)偏移。最高的峰(强度100%)以黑体字母示出。热分析富马酸盐形式a的dsc曲线显示出在231.3℃处存在一个吸热峰,δhfus为106.9j/g(图34)。形式a在25与150℃之间具有0.2%的重量损失。化合物a对甲苯磺酸盐形式a制备根据实施例5制备盐。xrpd对甲苯磺酸盐形式a的表征在图35和表21中描绘。表21.对甲苯磺酸盐形式a的xrpd峰*使用zbg或玻璃板通常引入正样品高度位移并导致2θ值的小(0.05°至0.2°)偏移。最高的峰(强度100%)以黑体字母示出。热分析对甲苯磺酸盐形式a的dsc曲线显示出在239.6℃处存在一个吸热峰,δhfus为38.5j/g(图36)。形式a在25与150℃之间具有0.04%的重量损失。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 作为AXL抑制剂的杂环化合物的制作方法与工艺
- HPTP‑β抑制剂的制作方法与工艺
- 作为LSD1抑制剂的环丙胺的制作方法与工艺
- KRAS G12C的抑制剂的制作方法与工艺
- 调节雌激素受体突变体的方法和组合物与流程
- 作为PI3K抑制剂的吡啶并[1,2‑A]嘧啶酮类似物的制作方法与工艺
- 作为PI3K抑制剂的吡啶并[1,2‑A]嘧啶酮类似物的制作方法与工艺
- 包含Ⅲ类受体酪氨酸激酶抑制剂和烷基化组蛋白‑脱乙酰酶抑制剂融合分子EDO‑S101的药物组合及其在治疗癌症中的用途的制作方法与工艺
- 包含Ⅲ类受体酪氨酸激酶抑制剂和烷基化组蛋白‑脱乙酰酶抑制剂融合分子EDO‑S101的药物组合及其在治疗癌症中的用途的制作方法与工艺
- 新型雌激素相关受体α抑制剂及其医学用途的制作方法与工艺