一种苯并呋喃类衍生物、其制备方法和应用与流程
本发明属于医药领域,具体而言,本发明涉及苯并呋喃类衍生物或其可药用盐、所述衍生物的制备方法、包含所述衍生物的药物组合物、以及所述衍生物和药物组合物在制备抗心律失常药物和治疗相关疾病中的用途。
背景技术:
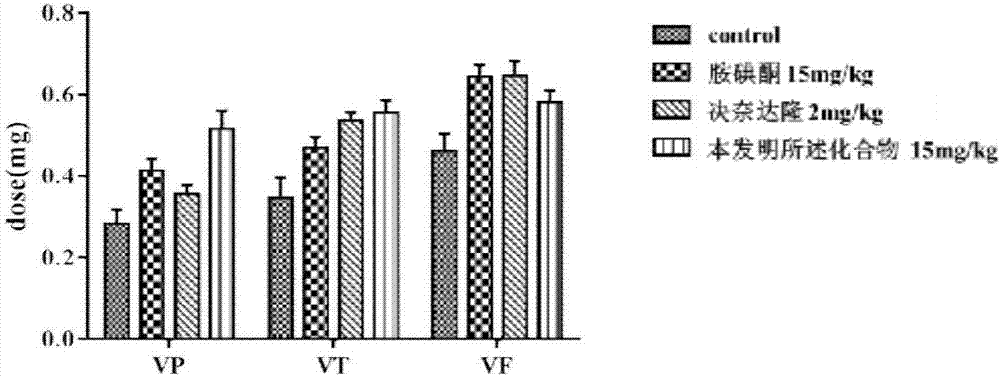
:胺碘酮属于苯并呋喃类衍生物,用于室性心律失常、心房颤动等的预防治疗,作为抗心律失常药物应用于临床已40余年。在CAST实验后,针对胺碘酮进行了13个多达近万人的临床实验,发现胺碘酮不增加也不减少有器质性心脏病包括心衰、心梗的患者的死亡率,只减少心律失常性死亡率。其对器质性心脏病患者的安全性使得胺碘酮在临床上得以保留。但是胺碘酮在使用后血药浓度迅速降低,大量分布于组织中,个体差异的存在导致不同患者使用胺碘酮时生效时间难以预测,需要随时监测,增大了临床使用的难度。同时由于胺碘酮分子结构中含有2个碘原子,占其相对分子质量的37.2%,治疗2~3个月后,约5%~28%的患者出现甲状腺功能障碍,肺纤维化、眼睛、皮肤等部位副作用发生的风险也随着长期用药而增加,且因其体内蓄积作用使得其不良反应难以消除。决奈达隆(Dronedarone,SR33589)是法国赛诺菲-安万特公司开发的Ⅲ类抗心律失常药,2009年7月1日在美国批准上市,2009年12月16日获欧盟EMEA批准,适用于心房纤颤(AF)和心房扑动症(AFL)患者的心律控制、维持窦性心律和减慢室性心律,临床主要用于治疗心律失常。决奈达隆是结构与胺碘酮(Amiodarone)类似的苯并呋喃的衍生物,具有与胺碘酮类似的电生理作用,同时去掉了碘的毒副作用,改善了药代动力学。但是与胺碘酮相比决奈达隆负性肌力作用较强,增加了心衰病人和器质性心脏病人使用的死亡率,且上市后出现严重肝功能损害。在本公司申请的专利《苯并呋喃类衍生物、其制备方法和应用》(申请号:201410267113.8)中描述了一系列新型苯并呋喃类衍生物,水溶性、代谢稳定性优于决奈达隆,且作用强度优于胺碘酮。在此基础上我们对化合物结构进行了进一步修饰,研究结果表明,本发明所述化合物抗心律失常作用优于胺碘酮,体外稳定性优于决奈达隆,对心脏的负性肌力作用与胺碘酮相当,显著优于决奈达隆,有望成为兼具有效性和安全性的新型抗心律市场药物。技术实现要素:本发明的第一个目的在于提供式I所示的化合物,或其药学上可接受的盐、溶剂化物、水合物、药学上可接受的前药,式I所示化合物的结构式如下:本发明所述式I结构的化合物为2-丁基-3-[3-甲磺酰基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-甲磺酰氨基苯并呋喃。本发明所述式I化合物的药学上可接受的盐,为式Ⅰ所示的化合物与有机酸或无机酸所成的盐。式Ⅰ所代表的化合物可以与无机酸形成药用盐,例如硫酸盐、磷酸盐、盐酸盐、氢溴酸盐;也可以与有机酸形成药用盐,例如醋酸盐、草酸盐、柠檬酸盐、琥珀酸盐、葡萄糖酸盐、酒石酸盐、对甲苯磺酸盐、苯磺酸盐、甲磺酸盐、苯甲酸盐、乳酸盐、马来酸盐等。本发明的第二个目的在于提供式Ⅰ所代表的苯并呋喃类衍生物或其可药用盐、溶剂化物、水合物、药学上可接受的前药的制备方法。本发明所述的制备方法,包括以下步骤:(1)使式Ⅱ所示化合物与硝酸反应生成式Ⅲ所示化合物:(2)使式Ⅲ所示化合物在碱作用下与1-溴-3-氯丙烷反应生成式Ⅳ所示化合物:(3)使式Ⅴ所示化合物在碱的作用下与哌啶反应生成式Ⅵ所示化合物:(4)使式Ⅴ所示化合物在还原剂条件下生成式Ⅵ所示化合物:(5)使式Ⅵ所示化合物在碱性条件下与甲磺酰氯反应生成化合物Ⅰ:(6)使化合物Ⅰ与相应的酸反应生成式Ⅷ所示化合物盐,其中A为酸根:上述制备过程中,所用到的化合物Ⅱ-Ⅰ都属于现有产品,都可以在市场购买到。优选的,本发明的制备方法,包括以下步骤:1)2-丁基-3-(3-硝基-4-羟基苯甲酰基)-5-硝基苯并呋喃的制备:将2-丁基-3-(4-羟基苯甲酰基)-5-硝基苯并呋喃(5.00g,14.7mmol)溶于100mL二氯甲烷中,滴加适量浓硝酸,室温反应,加水和二氯甲烷萃取,二氯甲烷相浓缩,得黄色固体,2)2-丁基-3-[3-硝基-4-(3-氯丙氧基)苯甲酰基]-5-硝基苯并呋喃的制备:将2-丁基-3-(3-硝基-4-羟基苯甲酰基)-5-硝基苯并呋喃(3.7g,9.64mmol)加入50mlDMF中,加入碳酸钾(4.0g,28.9mmol),碘化钾(0.4g,2.41mmol),和1-溴-3-氯丙烷(6.83g,43.4mmol),加热反应,将反应液冷至室温,过滤,滤液加入乙酸乙酯和水萃取,分液,乙酸乙酯相用水和饱和食盐水洗涤,浓缩,过滤,得固体,3)2-丁基-3-[3-硝基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-硝基苯并呋喃的制备:将2-丁基-3-[3-硝基-4-(3-氯丙氧基)苯甲酰基]-5-硝基苯并呋喃(1.5g,3.26mmol)溶于40ml乙腈,加入哌啶(2.8g,32.5mmol),碳酸钾(1.2g,8.70mmol),碘化钾(0.15g,0.6mmol),加热回流搅拌,冷至室温,浓缩,加入乙酸乙酯和水萃取,分液,有机相用水和饱和食盐水洗涤,浓缩,硅胶柱分离纯化,得黄色油状物,4)2-丁基-3-[3-氨基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-氨基苯并呋喃的制备:将2-丁基-3-[3-硝基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-硝基苯并呋喃(1.0g,1.96mmol)溶于50mlTHF中,加入适量Pd/C,置换氢气,室温搅拌4h,过滤,浓缩,得黄色油状液体,柱分离,得纯品,5)2-丁基-3-[3-甲磺酰氨基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-甲磺酰氨基苯并呋喃的制备:将2-丁基-3-[3-氨基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-氨基苯并呋喃(1.0g,2.22mmol)溶于45mlDCM中,加入碳酸氢钠(1.12g,13.3mmol),室温搅拌下滴加甲基磺酰氯(0.76g,6.67mmol)的DCM溶液,加水淬灭,分液,DCM相用水洗2次,浓缩,剩余物经硅胶柱分离纯化,得较纯的产品,6)2-丁基-3-[3-甲磺酰氨基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-甲磺酰氨基苯并呋喃盐酸盐的的制备:将2-丁基-3-[3-甲磺酰氨基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-甲磺酰氨基苯并呋喃(0.43g,0.71mmol)溶于乙腈中,滴加适量盐酸溶液,室温下搅拌,将反应液减压浓缩得黄色固体。本发明的第三个目的在于提供一种药物组合物,含有至少一种式Ⅰ所代表的苯并呋喃类衍生物,或其药学上可接受的盐、溶剂化物、水合物、药学上可接受的前药作为药物活性成分。根据需要,本发明的药物组合物还可以加入一种或多种药学上可接受的载体或赋形剂。本发明的药物组合物,式Ⅰ所代表的苯并呋喃类衍生物,或其药学上可接受的盐、溶剂化物、水合物、药学上可接受的前药所占重量百分比可以是0.1-99.9%,其余为药物可接受的载体。本发明的药物组合物可以制备成任何可药用的剂型,这些剂型包括:片剂、糖衣片剂、薄膜衣片剂、肠溶衣片剂、胶囊剂、硬胶囊剂、软胶囊剂、口服液、口含剂、颗粒剂、冲剂、丸剂、散剂、膏剂、丹剂、混悬剂、粉剂、溶液剂、注射剂、栓剂、软膏剂、硬膏剂、霜剂、喷雾剂、滴剂、贴剂。本发明的制剂,优选的是口服剂型,如:胶囊剂、片剂、口服液、颗粒剂、丸剂、散剂、丹剂、膏剂等。本发明给药途径可以是口服、非肠道或局部给药,优选口服和注射形式给药。适于药用的口服给药制剂可以是片剂、胶囊、颗粒剂或其它适于药用的液体形式的制剂如溶液、乳液、悬浮剂等。优选的口服制剂是片剂,并且所述片剂可以制成包衣、肠溶、缓释或定量释放的形式。本发明的药物组合物,其口服给药的制剂可含有常用的赋形剂,诸如粘合剂、填充剂、稀释剂、压片剂、润滑剂、崩解剂、着色剂、调味剂和湿润剂,必要时可对片剂进行包衣。适用的填充剂包括纤维素、甘露糖醇、乳糖和其它类似的填充剂。适宜的崩解剂包括淀粉、聚乙烯吡咯烷酮和淀粉衍生物,例如羟基乙酸淀粉钠。适宜的润滑剂包括,例如硬脂酸镁。适宜的药物可接受的湿润剂包括十二烷基硫酸钠。可通过混合,填充,压片等常用的方法制备固体口服组合物。进行反复混合可使活性物质分布在整个使用大量填充剂的那些组合物中。口服液体制剂的形式例如可以是水性或油性悬浮液、溶液、乳剂、糖浆剂或酏剂,或者可以是一种在使用前可用水或其它适宜的载体复配的干燥产品。这种液体制剂可含有常规的添加剂,诸如悬浮剂,例如山梨醇、糖浆、甲基纤维素、明胶、羟乙基纤维素、羧甲基纤维素、硬脂酸铝凝胶或氢化食用脂肪,乳化剂,例如卵磷脂、脱水山梨醇一油酸酯或阿拉伯胶;非水性载体(它们可以包括食用油),例如杏仁油、分馏椰子油、诸如甘油的酯的油性酯、丙二醇或乙醇;防腐剂,例如对羟基苯甲酯或对羟基苯甲酸丙酯或山梨酸,并且如果需要,可含有常规的香味剂或着色剂。对于注射剂,制备的液体单位剂型含有本发明的活性物质和无菌载体。根据载体和浓度,可以将此化合物悬浮或者溶解。溶液的制备通常是通过将活性物质溶解在一种载体中,在将其装入一种适宜的小瓶或安瓿前过滤消毒,然后密封。辅料例如一种局部麻醉剂、防腐剂和缓冲剂也可以溶解在这种载体中。为了提高其稳定性,可在装入小瓶以后将这种组合物冰冻,并在真空下将水除去。本发明的药物组合物,在制备成药剂时可选择性的加入适合的药物可接受的载体,所述药物可接受的载体选自:甘露醇、山梨醇、焦亚硫酸钠、亚硫酸氢钠、硫代硫酸钠、盐酸半胱氨酸、巯基乙酸、蛋氨酸、维生素C、EDTA二钠、EDTA钙钠,一价碱金属的碳酸盐、醋酸盐、磷酸盐或其水溶液、盐酸、醋酸、硫酸、磷酸、氨基酸、氯化钠、氯化钾、乳酸钠、木糖醇、麦芽糖、葡萄糖、果糖、右旋糖苷、甘氨酸、淀粉、蔗糖、乳糖、甘露糖醇、硅衍生物、纤维素及其衍生物、藻酸盐、明胶、聚乙烯吡咯烷酮、甘油、土温80、琼脂、碳酸钙、碳酸氢钙、表面活性剂、聚乙二醇、环糊精、β-环糊精、磷脂类材料、高岭土、滑石粉、硬脂酸钙、硬脂酸镁等。本发明式Ⅰ所代表的苯并呋喃类衍生物,或其药学上可接受的盐、溶剂化物、水合物、药学上可接受的前药,可以单独或以药物组合物的形式给药。本发明药物组合物可根据给药途径配成各种适宜剂型。使用一种或多种生理学上可接受的载体,包含赋形剂和助剂,它们有利于将活性化合物加工成可以在药学上使用的制剂。适当的制剂形式取决于所选择的给药途径,可以按照本领域熟知的常识进行制备。本发明的第四个目的在于提供式Ⅰ所代表的苯并呋喃类衍生物,或其药学上可接受的盐、溶剂化物、水合物、药学上可接受的前药在制备抗心律失常的药物中的应用。本发明所述心律失常所对应的相关适应症包括:阵发性或持续性心房颤动(AF)或心房扑动(AFL)、近期AF/AFL发作、伴心血管风险症(包括高血压、糖尿病、既往心血管意外、左心房直径≥50mm或左心室射血分数[LVEF]<40%)、窦性心律或心律可复律症或其组合。本发明的第五个目的在于提供以苯并呋喃类衍生物或其可药用盐作为活性成分的药物组合物在制备抗心律失常的药物中的应用。本发明所述的苯并呋喃类衍生物或其可药用盐,其水溶性明显优于盐酸决奈达隆,且体外稳定性及抗心律失常作用均优于盐酸决奈达隆。本发明相对于现有产品而言,大大增强药物活性,提高抗心律失常的治疗效果,增强药物的稳定性,降低副作用,而且合成过程操作也更加简单,大大降低成本,适合大规模生产。附图说明图1、各组样本给药后与给药前Control组的数据图2、各种化合物对哇巴因导致心律失常发生所需的剂量具体实施方式下面通过具体的实施方式对本发明的技术方案作进一步的说明,其中例举的实施例是对本发明的说明,而不以任何方式限制其保护范围。实施例12-丁基-3-(3-硝基-4-羟基苯甲酰基)-5-硝基苯并呋喃将2-丁基-3-(4-羟基苯甲酰基)-5-硝基苯并呋喃(5.00g,14.7mmol)溶于100mL二氯甲烷中,滴加浓硝酸(5ml,72.5mmol),室温反应40min,加水和二氯甲烷萃取,二氯甲烷相浓缩,得黄色固体5.5g,收率97%。实施例22-丁基-3-[3-硝基-4-(3-氯丙氧基)苯甲酰基]-5-硝基苯并呋喃将2-丁基-3-(3-硝基-4-羟基苯甲酰基)-5-硝基苯并呋喃(3.7g,9.64mmol)加入50mlDMF中,加入碳酸钾(4.0g,28.9mmol),碘化钾(0.4g,2.41mmol),和1-溴-3-氯丙烷(6.83g,43.4mmol),70℃反应3h,将反应液冷至室温,过滤,滤液加入乙酸乙酯和水萃取,分液,乙酸乙酯相用水和饱和食盐水洗涤,浓缩,用PE:EA=1:1打浆,过滤,得固体3.0g,收率67.5%。实施例32-丁基-3-[3-硝基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-硝基苯并呋喃将2-丁基-3-[3-硝基-4-(3-氯丙氧基)苯甲酰基]-5-硝基苯并呋喃(1.5g,3.26mmol)溶于40ml乙腈,加入哌啶(2.8g,32.5mmol),碳酸钾(1.2g,8.70mmol),碘化钾(0.15g,0.6mmol),加热85℃回流搅拌4h,冷至室温,浓缩,加入50ml乙酸乙酯和50ml水萃取,分液,有机相用水(50ml×2)和饱和食盐水(50ml×2)洗涤,浓缩,硅胶柱分离纯化(洗脱剂,二氯甲烷:甲醇=20:1),得黄色油状物1.2g,收率为70%,LC-MS:m/z510.2[M+H]+。实施例42-丁基-3-[3-氨基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-氨基苯并呋喃将2-丁基-3-[3-硝基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-硝基苯并呋喃(1.0g,1.96mmol)溶于50mlTHF中,加入Pd/C(0.29g),置换氢气,室温搅拌4h,过滤,浓缩,得黄色油状液体,柱分离,得0.85g纯品,收率96.5%,LC-MS:m/z450.3.[M+H]+。实施例52-丁基-3-[3-甲磺酰氨基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-甲磺酰氨基苯并呋喃将2-丁基-3-[3-氨基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-氨基苯并呋喃(1.0g,2.22mmol)溶于45mlDCM中,加入碳酸氢钠(1.12g,13.3mmol),室温搅拌下滴加甲基磺酰氯(0.76g,6.67mmol)的DCM溶液,反应10h,加水淬灭,分液,DCM相用水洗2次,浓缩,剩余物经硅胶柱分离纯化(洗脱剂,二氯甲烷∶甲醇=50∶1,v∶v),得0.76g较纯的产品,收率56.5%,LC-MS:m/z606.2[M+H]+。实施例62-丁基-3-[3-甲磺酰氨基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-甲磺酰氨基苯并呋喃盐酸盐将2-丁基-3-[3-甲磺酰氨基-4-[3-(哌啶-1-基)丙氧基]苯甲酰基]-5-甲磺酰氨基苯并呋喃(0.43g,0.71mmol)溶于10ml乙腈中,滴加6mol/L的盐酸溶液0.2ml,室温下搅拌30min,将反应液减压浓缩得0.44g黄色固体,收率为96%。1HNMR(400MHz,DMSO-d6):δ=0.80(t,J=7.2Hz,3H),1.22-1.24(m,2H),1.38-1.40(m,2H),1.48-1.51(m,4H),1.63-1.65(m,2H),1.96-1.98(m,2H),2.36-2.37(m,4H),2.46(t,J=7.2Hz,2H),2.81(t,J=7.2Hz,2H),2.90(s,3H),2.97(s,3H),4.16(t,J=6.0Hz,2H),7.20-7.23(m,2H),7.31(d,J=2.0Hz,1H),7.61-7.65(m,2H),7.73(d,J=2.0Hz,1H),9.60(s,1H)。测试例生物学评价测试例1本发明化合物对hERG钾通道的抑制作用实验步骤:(1)收集的细胞(重组HEK293细胞系表达人hERG(ether-a-go-gorelatedgene)钾通道)悬液置于细胞池中,每30s吹吸细胞一次,以避免细胞沉降或成团。机械臂自动注入细胞内液,细胞外液并将细胞悬液注入封接芯片。细胞在负压得吸引下随机附着在孔上,然后通过抽吸使附着在孔上的膜片破裂,形成全细胞记录模式。(2)全细胞膜片钳记录按照patchmaster制定的标准程序完成。当全细胞记录稳定后开始给药(实验组为本发明化合物;对照组为胺碘酮),每个药物浓度作用至稳定后检测下一个浓度,在记录期间独立重复检测3个细胞。(3)全细胞膜片钳记录全细胞hERG钾电流记录方法如下:钳制电压由-80mV除极至+40mV维持500毫秒,然后迅速保持在-40mV维持500毫秒,记录尾电流,并每隔10秒重复采集数据。数据由HEKAEPC-10Quatro放大器进行采集并储存于PatchMaster软件中。所有电生理实验均在室温下(25℃)。质量控制:(1)实验的整个过程中,所有数据符合以下标准方被采纳:(2)整个实验中破膜前封接电阻Rseal>1GΩ。(3)电流衰减在阴性对照组中低于1%/min(4)漏电流小于100pA。(5)整个实验中串联电阻Rs<20MΩ。(6)给药前电流大于400pA。数据分析:(1)所有数据以n个细胞的均值±标准差表示。(2)标准化的电流幅值通过以下方程进行拟合:1(1-(c(IC50)-1)h)-1。方程中c为药物浓度,IC50为最大效应50%的药物浓度,h为希尔系数。拟合通过NanionTechnologies整合的数据分析软件包完成。实验结果:下表概述化合物对hERG钾通道的抑制作用。化合物本发明化合物hERGIC506.3μM±715.5nM测试例2本发明所述化合物对豚鼠离体心脏收缩力的作用决奈达隆,胺碘酮属于III类抗心律失常药物,胺碘酮类药物对于心脏的离子通道具有广泛的抑制作用,文献报道决奈达隆和胺碘酮均可以浓度依赖性地抑制心脏钠通道,钙通道,以及多种钾通道,如IKr,IKs,Ito,IK1等等。文献报道决奈达隆具有负性肌力作用,严重影响决奈达隆的临床使用范围。本研究中,我们利用langendorff离体心脏灌流技术,检测本发明所述化合物及胺碘酮和决奈达隆对心脏的负性肌力作用。从而评价和比较本发明所述化合物以及胺碘酮和决奈达隆对正常离体心肌收缩力影响的差异。实验步骤:豚鼠体重350-400g,雌雄不限。腹腔注射戊巴比妥钠(30mg·kg-1)、肝素(250u·kg-1)。麻醉生效后,先剪开腹腔,然后经膈肌于胸腔两侧剪开胸腔,止血钳夹住胸前壁,翻向头部,完全暴露出心脏,用拇指与食指轻捏心脏底部(尽量避免挤压心脏),轻轻提起,剪下心脏,尽量保留较长的主动脉。将心脏迅速置室温(18-22℃)的清洗液中,迅速行主动脉插管,恢复冠脉灌流。心脏悬垂于Langendorff灌流装置上,以60mmHg压力行心脏逆行灌流。灌流液为氧合的Krebs-Henseleit缓冲液,温度保持在37℃。在左心房处剪一小口,将带有乳胶水囊的测压管经二尖瓣口插入左心室,连接压力换能器,调节囊内压为10mmHg。MedLab3000生理信号采集分析系统记录左心室内压力变化,分析获取左心室收缩压力(leftventricularsystolicpressure,LVSP,单位mmHg)、左心室舒张末期压力(leftventricularend-diastolicpressure,LVEDP,单位mmHg)、左心室内压最大上升速率(+dp/dtmax,单位mmHg/s)、左心室内压最大下降速率(-dp/dtmax,单位mmHg/s)与心率(heartrate,HR,单位beats/s)等数据。稳定20分钟后给药(本发明所述化合物、胺碘酮及决奈达隆),每个浓度给药至少40分钟,记录给药前后心脏左室收缩压及最大上升速率,左室舒张期末压,收缩压,舒张压的改变。实验结果:1μM浓度作用下,本发明所述化合物对豚鼠心肌收缩力的负性肌力作用最小,与胺碘酮相当,决奈达隆有显著的负性肌力作用。下表概述化合物对豚鼠离体心脏收缩力的作用将各组样本给药后与给药前Control组的数据作比进行标准化处理后,实验结果见附图1所示。在另外一组测试中,将本发明的化合物与中国专利(201410267113.8)所对应的化合物进行比较,实验结果显示,本发明的化合物在心脏收缩力方面表现出更好的效果。测试例3化合物对哇巴因致家兔心律失常模型的作用本实验的目的是比较研究本发明所述化合物和胺碘酮静脉给药对哇巴因诱发兔心律失常模型的作用,从而评价本发明所述化合物的抗心律失常作用。实验步骤:实验动物实验前禁食12h,自由饮水,实验具体操作如下:(1)家兔称重,25%乌拉坦经耳缘静脉注射麻醉,并设置静脉留置针;(2)家兔仰卧固定于手术台上;(3)powerlab系统记录标准肢体II导联心电图(位置为右前肢,左后肢,左后肢);(4)连续监测心电图15min后通过静脉留置针一次性注射胺碘酮或受试药物5ml,空白对照组注射5%葡萄糖注射液5ml;(5)连续监测心电图30min后,连接注射泵,哇巴因溶液以静脉滴注的方式连续给药,给药速度为15μg/min(0.1ml/min);(6)连续监测心电图并记录出现室性早搏(VP)、室性心动过速(VT)、心室纤颤(VF)及心跳停止的时间及哇巴因用量。实验结果:下表概述化合物对哇巴因致心律失常作用剂量的影响。化合物剂量室性早搏室性心动过速室颤对照组-0.283±0.03460.349±0.04690.462±0.0415本发明化合物15mg/kg0.515±0.0770**0.555±0.05200.580±0.0527胺碘酮15mg/kg0.413±0.0295***0.47±0.0256*0.643±0.0302***决奈达隆2mg/kg0.371±0.0662*0.576±0.07380.683±0.0822**p<0.05,**p<0.01,***p<0.001,vs.对照组由上表可看出本发明所述化合物显著增加哇巴因导致心律失常发生所需的剂量,作用强于同剂量胺碘酮。其作用强度见附图2所示。测试例4体外肝微粒体代谢稳定性研究实验步骤:将含有浓度为1.27mg/mL的人肝微粒体的磷酸缓冲液(0.05M,Ph=7.4)加入1.1mL的试管中,加入2.5μL的待测化合物(本发明所述化合物及决奈达隆),于37℃中预孵5min,加入50μLNADPH溶液,待测化合物的终浓度为1μM(1%DMSO),温孵液的总体积为250μL。在0、15、30、45和60分钟时从反应体系中取出15μL的等分试样,加入200μL的甲醇/乙腈(1:1)终止反应,得到的混合物3400rpm离心15min,上清液用于LC-MS/MS分析。LC分析采用的仪器和条件为:色谱柱:Kinetex2.6uC18100Acolumn(3.0mm×30mm)流动相:0.1%甲酸水溶液(A)和0.1%甲酸-乙腈(B);洗脱程序为0~0.5min,维持流动相B为5%;0.5~1.0min,流动相B为5%到95%;1.0~1.5min,维持流动相B为95%;1.5~2.0min,平衡至流动相B为5%。运行时间为2min,流速为1mL/min,上样体积为5μL。MS分析采用的仪器和条件为:API4000QTrap电离模式:ESI扫描类型:MRM实验结果:在存在NADPH的人肝微粒体体系中,本发明所述化合物的体外稳定性强于决奈达隆,结果如下表所示:当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种具有抗氧化活性的芳基苯并呋喃类化合物及其制备方法和应用与流程
- 一种含苯并双环酮与乙氧磺隆的除草组合物及其应用的制造方法与工艺
- 一种6‑氯呋喃[3,2‑B]吡啶的合成方法与流程
- 一类苯并吡喃类化合物及其应用的制造方法与工艺
- 一种以二苯并环庚烯为核心的化合物及其在有机电致发光器件上的应用的制造方法与工艺
- 一种含苯并双环酮与乙草胺的除草组合物及其应用的制造方法与工艺
- 一种含苯并双环酮与噁草酮的除草组合物及其应用的制造方法与工艺
- 分解二噁英前体的催化剂筛选系统及其使用方法与流程
- 一种无卤热固性树脂组合物以及含有它的预浸料、层压板和印制电路板的制造方法与工艺
- 一种以二联二苯并五元杂环为骨架的有机化合物及其在OLED上的应用的制造方法与工艺