狂犬病毒DRV-AH08株的先导RNA及其在制备预防与治疗狂犬病毒的药品中的应用的制作方法
本发明涉及公共卫生和预防兽医学领域,具体地指一种狂犬病毒DRV-AH08株的先导RNA及其在制备预防与治疗狂犬病毒的药品中的应用。
背景技术:
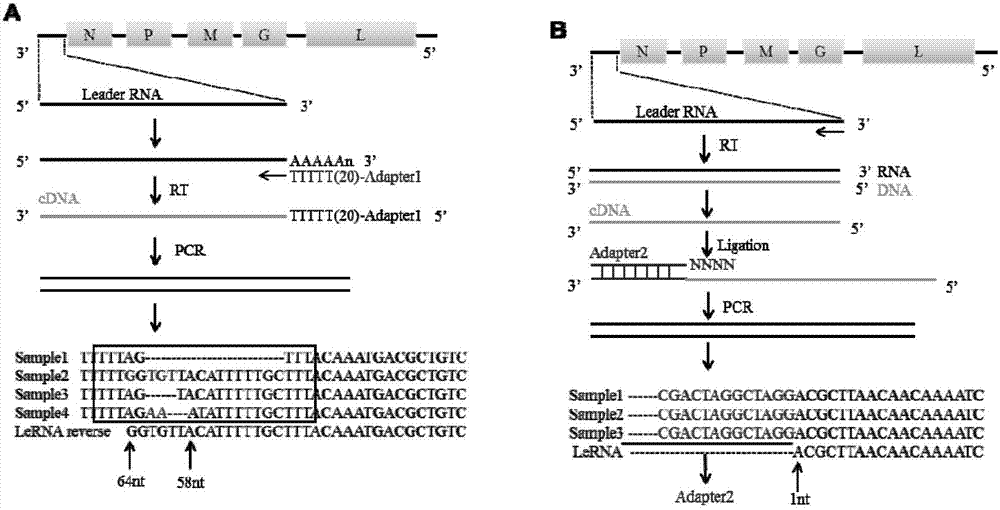
:狂犬病是一种人兽共患传染病,所有温血动物都能感染,至今仍然在全世界广泛流行。2002~2007年,全国狂犬病死亡11198例,占同期各种传染病病死数的30%,高居所有法定传染病病死数之首。因此狂犬病与艾滋病、病毒性肝炎、结核病一样,已成为危害我国人口健康与公共卫生安全的重大传染病。狂犬病一旦发病,其致死率几乎为100%,因而控制狂犬病的主要手段是预防。免疫预防分为暴露前和暴露后免疫。暴露前免疫是给经常与狂犬病病毒接触的人接种疫苗。暴露后免疫是给被患狂犬病动物或者被疑似狂犬病动物咬伤,舔伤,抓伤的人接种疫苗。对伤势过重者还要用抗狂犬病血清进行治疗。但是暴露后免疫和治疗一定要尽早、及时,一旦出现临床症状,就预示着暴露后免疫和治疗的失败和患者的死亡。狂犬病毒因其高致死率,对公共卫生安全危害极大。狂犬病毒感染后病毒通过神经进入血脑屏障,神经细胞坏死和凋亡不明显,病理变化不典型,甚至有的病例其脑组织标本无炎症反应,狂犬病的发病机理始终不清楚,为发病后治疗带来极大困难,目前狂犬病暴露后的治疗只有使用含有中和抗体的抗血清这一种方法,而抗血清价格昂贵,而且可能产生过敏性休克和血清病等不良反应,这使得抗血清的使用有一定的局限性。技术实现要素:本发明目的是针对现有技术存在的缺陷,提供了一种狂犬病毒DRV-AH08株的先导RNA及其在制备预防与治疗狂犬病毒的药品中的应用。为实现上述目的,本发明提供的一种狂犬病毒DRV-AH08株的先导RNA,所述先导RNA的核苷酸序列为下述长短不一的RNA核苷酸序列中任意一种,分别为先导RNA1的核苷酸序列如SEQIDNo.1所示,先导RNA2的核苷酸序列如SEQIDNo.2所示,先导RNA3的核苷酸序列如SEQIDNo.3所示,先导RNA4的核苷酸序列如SEQIDNo.4所示,先导RNA5的核苷酸序列如SEQIDNo.5所示,先导RNA6的核苷酸序列如SEQIDNo.6所示,先导RNA7的核苷酸序列如SEQIDNo.7所示。先导RNA(LeaderRNA)是狂犬病毒感染宿主后产生的一条单股正链的非编码小RNA,长度约64nt,先导RNA既不加帽也无polyA尾巴,是狂犬病毒感染后最早的转录产物。本发明利用先导RNA这一特性,在狂犬病毒感染后,用先导RNA来进行治疗,发挥类似于RNAi的作用来抑制狂犬病毒的复制,也尚未有相关报道。本发明还提供了一种重组真核表达载体AAV-U6,所述重组真核表达载体AAV-U6是在真核表达载体AAV的NotI酶切位点之间插入U6-BbsI-CMV-mcherry片段得到的,其中,U6-BbsI-CMV-mcherry片段的核苷酸序列如SEQIDNo.8所示。上述重组真核表达载体AAV-U6的构建方法,包括以下步骤:1)PCR扩增U6启动子:以PX335质粒(购买于Addgene,编号:#61515)为模板设计引物对,同时下游引物的末端带有与CAGEnhancer序列互补的一段同源臂),其中,引物对为:U6-F-NotI:5’-AAATATGCGGCCGCGAGGGCCTATTTCCCATGATT-3’,NotIU6-R-BbsI:5’-CATTTACCGTAAGTTATGTAAAAAAGGTCTTCTCGAAGACCC-3’;BbsI反向BbsI进行PCR扩增,纯化得到NotI-U6-BbsI-BbsI-TTTTT片段,其核苷酸序列如SEQIDNo.9所示;2)PCR扩增mCherry表达盒:以pmCherry(本实验室保存)为模板设计引物对,mCherry核苷酸序列如SEQIDNo.10所示,其中,上游引物带有与NotI-U6-BbsI-BbsI-TTTTT序列末端相同的一段同源臂,下游引物带有NotI酶切位点;使用的引物序列如下:CMV-CAG-F-BbsI:5’-GGGTCTTCGAGAAGACCTTTTTTACATAACTTACGGTAAATG,反向BbsIBbsICMV-terminator-R-NotI:5’-AAATATGCGGCCGCTAAGATACATTGATGAGTTTGG-3’;NotIPCR扩增纯化得到CAG-CMV-mCherry-Terminator-NotI片段,其核苷酸序列如SEQIDNo.11所示;3)融合PCR扩增U6-BbsI-CMV-mCherry;以NotI-U6-BbsI-BbsI-TTTTT片段和CAG-CMV-mCherry-Terminator-NotI片段作为模板进行融合PCR;其中,引物对为:U6-F-NotI:5’-AAATATGCGGCCGCGAGGGCCTATTTCCCATGATT-3’,NotICMV-terminator-R-NotI:5’-AAATATGCGGCCGCTAAGATACATTGATGAGTTTGG-3’;NotIPCR扩增纯化得到U6-BbsI-CMV-mCherry片段,其核苷酸序列如SEQIDNo.8所示;4)酶切:NotI酶切载体pAAV和片段U6-BbsI-CMV-mCherry,酶切后纯化线性化的载体和片段;5)连接:用T4连接酶连接线性化的载体和片段,并转化大肠杆菌感受态Stbl3,挑取克隆并提质粒测序,鉴定正确即得到重组真核表达载体AAV-U6。在上述技术方案中,所述步骤1)中,PCR反应体系如下:PCR的条件如下:所述步骤2)中,PCR反应体系如下:CMV-CAG-F-BbsI:5’-GGGTCTTCGAGAAGACCTTTTTTACATAACTTACGGTAAATG-3’,反向BbsIBbsICMV-terminator-R-NotI:5’-AAATATGCGGCCGCTAAGATACATTGATGAGTTTGG-3’;NotIPCR的条件如下:所述步骤3)中,PCR反应体系如下:PCR的条件如下:本发明还提供了一种含有先导RNA对应的双链DNA序列的重组真核表达载体AAV-U6,所述先导RNA对应的双链DNA序列插入到重组真核表达载体AAV-U6的BbsI酶切位点之间。本发明还提供了一种含有上述重组真核表达载体AAV-U6的大肠杆菌Stbl3表达菌株。本发明提供了一种上述狂犬病毒DRV-AH08株的先导RNA在制备治疗和预防狂犬病的药品中的应用。本发明还提供了一种含有先导RNA对应的双链DNA序列的重组真核表达载体AAV-U6在制备治疗和预防狂犬病的药品中的应用。本发明还提供了一种含有重组真核表达载体AAV-U6的大肠杆菌Stbl3表达菌株在制备治疗和预防狂犬病的药品中的应用。本发明的有益效果在于:本发明采用类腺病毒载体在小鼠脑内过表达先导RNA,然后小鼠经脑感染狂犬病毒DRV-AH08株,后经脑切片染色观察发现过表达先导RNA能抑制狂犬病毒复制,并且用体外转录的方式获得纯的先导RNA,在SK-N-SH细胞系上转染先导RNA进入细胞,再感染狂犬病毒,经狂犬病毒染色观察发现病毒的复制被抑制。同时本发明也在SK-N-SH细胞系上感染狂犬病毒后再转染先导RNA,发现先导RNA仍然能抑制狂犬病毒的复制,在活体动物试验中,感染了狂犬病毒的小鼠,经注射能过表达先导RNA的类腺病毒,可以显著的增加小鼠的存活率。利用先导RNA对狂犬病进行暴露后治疗,在狂犬病的预防与治疗方面尚属先例,相对于抗血清,先导RNA价格较低,产生过敏性反应的可能性小,极具推广价值,这对于研究狂犬病暴露后治疗手段具有重要的意义和广阔的应用前景。附图说明图1为LeaderRNA转录起止位置的确定示意图;其中,图1A是转录终止位置确定,LeaderRNA转录终止的位置并不单一,大部分终止于离基因组3’末端64nt的位置;图1B是转录起始位置确定,LeaderRNA转录起始位置为基因组3’末端第一个碱基;图2为先导RNA在SK-N-SH细胞内过表达能抑制狂犬病毒DRV-AH08的复制图;图中,第一排为转染了对照RNA再感染狂犬病毒的细胞,在荧光条件下(左图)和明场条件下(右图)的成像。第二排为转染了先导RNA再感染狂犬病毒的细胞,在荧光条件下(左图)和明场条件下(右图)的成像。绿色表示狂犬病毒。图片中右下角白色矩形表示200μm比例尺。图3为先导RNA过表达载体pAAV-U6的结构示意图以及插入片段的位点(Insertionsite)和序列(Insert)示意图;图4为含有先导RNA对应的双链DNA序列的重组真核表达载体AAV-U6的图谱;图5为先导RNA在鼠脑内过表达能抑制狂犬病毒DRV-AH08的复制图;图中,图5A~C为为低倍镜下观察小鼠脑切片,图5A中左脑海马区的红色荧光为先导RNA过表达区域,右脑海马区的红色荧光为对照RNA过表达区域;图5B中绿色荧光表示狂犬病毒;图5C为A,B图叠加的效果图。图5D~I为鼠脑局部在高倍镜下观察的图片;图5D表示高倍镜下观察鼠脑左边海马区,红色为先导RNA过表达的区域;图5E表示与D图相同区域内狂犬病毒存在情况;图5F为D图与E图叠加的效果图,在先导RNA存在的区域(红色)狂犬病毒(绿色)的含量下降;图5G表示高倍镜下观察鼠脑右边海马区,红色为对照RNA过表达的区域;图5H表示与G图相同区域内狂犬病毒存在情况;图5I为H图与G图叠加的效果图,在对照RNA存在的区域(红色)狂犬病毒(绿色)的含量并没有减少,图中可见对照RNA存在的区域(红色)狂犬病毒(绿色)共同存在的细胞(黄色)。图6为SK-N-SH感染狂犬病毒DRV-AH08后过表达先导RNA能够抑制病毒复制;图6中,第一排为感染狂犬病毒后再转染对照RNA的细胞,在荧光条件下(左图)和明场条件下(右图)的成像;第二排为感染狂犬病毒后再转染先导RNA的细胞,在荧光条件下(左图)和明场条件下(右图)的成像。绿色表示狂犬病毒,图片右下角白色矩形表示200μm比例尺。图7为小鼠感染狂犬病毒DRV-AH08后过表达先导RNA能够降低小鼠死亡率的显示图。具体实施方式为了更好地解释本发明,以下结合具体实施例进一步阐明本发明的主要内容,但本发明的内容不仅仅局限于以下实施例。实施例1LeaderRNA1(先导RNA1)转录起始和终止位置的确定1、LeaderRNA1转录终止位置的确定1)用DRV-AH08经脑注射感染成年小鼠,小鼠发病后心脏灌注后取脑,提取脑组织RNA;2)用E.coliPoly(A)Polymerase(NEB)给RNA3’端加A,反应体系如下:反应程序:在温度为37℃条件下反应30~60min;3)用adapter1-d(T)将加A后的RNA反转成cDNA,adapter1-d(T)序列为:5’-ACTGTCAACTGGTGTCGTGGAGTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTVN-3’;反转反应体系如下:反应程序如下:室温10min42℃60min冰上2min4)反转后用一对引物进行扩增,上游引物来源于Adapter1,序列为:5’-ACTGTCAACTGGTGTCGTGGAGT-3’,下游引物来源于DRV-AH08基因组,序列为:5’-ACGCTTAACAACAAAATCAT-3’;扩增后的片段与pMD-18T连接,转化DH5α,挑菌30个,送测序,比对测序结果,与d(T)紧挨的序列即为leaderRNA转录终止的位置。2、LeaderRNA1转录起始位置的确定1)用DRV-AH08经脑注射感染成年小鼠,小鼠发病后心脏灌注后取脑,提取脑组织RNA;2)利用已知的转录终止位置,设计反转录引物,序列为:5’-GGTGTTACATTTTTGCTTTACAAA-3’,将脑组织RNA反转成cDNA;反转体系如下:反应程序:室温10min42℃60min冰上2min3)根据设计的adapter2序列合成单链核苷酸,序列分别为adapter2-1:5’-GATCCGAAGCAATACGACTAGGCTAGGNNNN-3’,adapter2-2:5’-CCTAGCCTAGTCGTATTGCTTCGGATC-3’;将合成的单链寡核苷酸成对变性、退火,退火之后形成具有粘性末端的DNA双链adapter2,粘性末端为NNNN,变性、退火体系如下:反应程序:4)反转得到的cDNA与退火成双链的adapter2连接,连接体系如下Adapter2退火产物1μlcDNA5μlT4连接酶(Takara)0.5μl10*T4连接酶缓冲液1μl总计7.5μl16℃孵育5-6h。5)连接后用一对引物进行扩增,上游引物来源于Adapter2,序列为:5’-GATCCGAAGCAATACGACTAGGCTA-3’,下游引物来源于LeaderRNA转录终止区域,序列为:5’-GGTGTTACATTTTTGCTTTAC-3’;扩增后的片段与pMD-18T连接,转化DH5α,挑菌30个,送测序,比对测序结果,与adapter2紧挨的序列即为leaderRNA转录起始的位置,先导RNA1的核苷酸序列如SEQIDNo.1所示。同理,通过上述方法得到其他先导RNA2~7核苷酸序列,如SEQIDNo.1~7所示实施例2先导RNA的过表达在体外和体内均能抑制狂犬病毒的复制1、先导RNA1的过表达在体外能抑制狂犬病毒的复制1)根据上述的先导RNA1核苷酸序列,利用体外转录的方法体外合成先导RNA,并纯化,分装后保存于-80℃。对照RNA购买于上海吉玛制药技术有限公司;2)SK-N-SH细胞用MEM培养基培养(含胎牛血清15%)。细胞长满后,胰酶消化、吹散,接种于24孔板,待细胞汇合度到70-80%后进行转染。3)按脂质体转染法,将先导RNA(40pmol)和对照RNA(40pmol)分别与脂质体混合,250μl转染液37℃孵育。每种RNA做6个复孔。4)转染后1h,按每孔0.01MOI感染DRV-AH08。37℃孵育6h后换维持液。5)转染后24小时,用4%多聚甲醛固定细胞,用FITC标记的狂犬病毒N蛋白单抗染色镜检。如图2所示:将先导RNA和对照RNA分别转染SK-N-SH细胞后,感染狂犬病毒DRV-AH08,24小时后固定细胞染色镜检。与转染了对照RNA的细胞相比,转染了先导RNA1的细胞狂犬病毒的复制明显减少。2、先导RNA1的过表达在体内能抑制狂犬病毒的复制1)根据上述的先导RNA1核苷酸序列,构建真核表达载体AAV-U6,AAV-U6载体示意图如图(3)所示:2)PCR扩增U6启动子,上游引物带有NotI酶切位点,下游引物带有两个反向的BbsI酶切位点,在BbsI酶切位点之后是连续的TTTTT(引物上为AAAAA),为U6启动子的终止子。同时下游引物的末端带有与CAGEnhancer序列互补的一段同源臂,扩增后得到NotI-U6-BbsI-BbsI-TTTTT片段(其核苷酸序列如SEQIDNo.9所示)并纯化。使用的引物序列如下:U6-F-NotI:5’-AAATATGCGGCCGCGAGGGCCTATTTCCCATGATT-3’;U6-R-BbsI:5’-CATTTACCGTAAGTTATGTAAAAAAGGTCTTCTCGAAGACCC-3’;PCR的条件如下:3)PCR扩增mCherry表达盒。上游引物带有与NotI-U6-BbsI-BbsI-TTTTT序列末端相同的一段同源臂,下游引物带有NotI酶切位点。扩增后得到CAG-CMV-mCherry-Terminator-NotI片段其核苷酸序列如SEQIDNo.11所示并纯化。使用的引物序列如下:CMV-CAG-F-BbsI:5’-GGGTCTTCGAGAAGACCTTTTTTACATAACTTACGGTAAATG-3’,CMV-terminator-R-NotI:5’-AAATATGCGGCCGCTAAGATACATTGATGAGTTTGG-3’;PCR的条件如下:4)融合PCR扩增U6-BbsI-CMV-mCherry。使用U6-F-NotI和CMV-terminator-R-NotI分别作为上下游引物,以NotI-U6-BbsI-BbsI-TTTTT片段和CAG-CMV-mCherry-Terminator-NotI片段作为模板,做融合PCR,获得U6-BbsI-CMV-mCherry片段并纯化。PCR的条件如下:5)分别用NotI酶切载体pAAV和片段U6-BbsI-CMV-mcherry,酶切后纯化线性化的载体和片段。6)用T4连接酶连接线性化的载体和片段,并转化大肠杆菌感受态Stbl3。7)挑取克隆并提质粒测序,鉴定正确;8)使用之前构建的真核表达载体AAV-U6,将先导RNA1插入AAV-U6载体中并包装成AAV病毒,在活体内过表达先导RNA1。同时用过表达其他非编码小RNA的AAV病毒作为对照。9)过表达载体构建方式如下:根据先导RNA和对照RNA序列在其正向寡核苷酸Forwardoligo5’端加上CACCG,在其逆向寡核苷酸Reverseoligo5’端加上AAAA,分别合成。如下:Forwardoligo:5’-CACCGNNNNNNNNNNNNNNNNNNN-3’,Reverseoligo:3’-CNNNNNNNNNNNNNNNNNNNAAAA-5’;将合成的两条互补的寡核苷酸序列成对变性、退火,退火之后形成具有粘性末端的DNA双链,可连入经Bbs1酶切线性化的AAV-U6真核表达载体中。变性、退火体系如下:反应程序:10)Bbs1酶切线性化AAV-U6真核表达载体;酶切体系和反应条件如下:37℃孵育3-4h,酶切完成后,琼脂糖凝脂电泳后,BioFluxR胶回收试剂盒回收,溶于30-40μlddH2O中。11)变性退火得到的先导RNA1、对照RNA双链寡聚核苷酸,分别与经Bbs1酶切线性化的质粒载体连接;反应体系如下:sgRNA退火产物1μlBbs1线性化载体50ngT4连接酶(Takara)0.5μl10xT4连接酶缓冲液1μl16℃孵育1-3h;12)将上述连接产物转化大肠杆菌感受态细胞(Stbl3),并涂布氨苄抗性平板(50μg/ml),并挑取单克隆菌落。13)DNA测序鉴定阳性克隆,得到AAV-U6-先导RNA1和AAV-U6-对照RNA的表达载体。2、小鼠实验1)使用三维立体脑定位注射仪将rAAV-U6-先导RNA1注射于小鼠左脑海马区内,再将rAAV-U6-对照RNA注射于同一只小鼠右脑海马区内注射后两周,分别经左右脑注射DRV-AH08病毒。2)待小鼠发病濒死时,麻醉小鼠,做心脏灌注,取鼠脑,并用4%多聚甲醛固定过夜。3)固定后的鼠脑用震荡切片机切片,厚度为30μm。分别用本实验室制作的狂犬病毒P蛋白单抗和购买的羊抗鼠二抗(绿色)孵育,孵育后于荧光显微镜下观察拍照如图5所示。在注射了rAAV-U6-先导RNA的鼠脑区域(左边海马区,红色),狂犬病毒的量(绿色)明显少于注射了rAAV-U6-对照RNA的鼠脑区域(右边海马区,红色),说明过表达先导RNA1能在活体水平显著抑制狂犬病毒的复制。实施例3先导RNA1对狂犬病毒的感染有潜在的治疗作用1、细胞水平的治疗1)根据上述的先导RNA1核苷酸序列,利用体外转录的方法体外合成先导RNA1,并纯化,分装后保存于-80℃。对照RNA购买于上海吉玛制药技术有限公司;2)SK-N-SH细胞用MEM培养基培养(含胎牛血清15%)。细胞长满后,胰酶消化、吹散,接种于24孔板,待细胞汇合度到70-80%后感染狂犬病毒;3)按每孔0.01MOI感染DRV-AH08,37℃孵育1h后转染RNA;4)按脂质体转染法,将先导RNA(40pmol)和对照RNA(40pmol)分别与脂质体混合,250μl转染液37℃孵育6h后换维持液,每种RNA做6个复孔;5)转染后24小时,用4%多聚甲醛固定细胞,用FITC标记的狂犬病毒N蛋白单抗染色镜检,如图6所示。细胞感染狂犬病毒后,转染了先导RNA1的细胞,狂犬病毒荧光斑的数量明显少于转让了对照RNA的细胞,说明狂犬病毒感染后,在细胞水平上,过表达先导RNA1仍然可以抑制狂犬病毒的进一步复制。2、活体水平的治疗1)根据上述的先导RNA1核苷酸序列,使用上述真核表达载体AAV-U6,在活体内过表达先导RNA1。同时用过表达其他非编码小RNA的AAV病毒作为对照。AAV-U6能表达红色荧光蛋白作为指示;2)小鼠经后肢肌肉感染狂犬病毒;1小时后将过表达先导RNA1的病毒AAV-U6-先导RNA1和过表达对照RNA的病毒AAV-U6-对照RNA注射进小鼠后肢肌肉,注射了狂犬病毒的位置附近。观察实验组和对照组的小鼠存活率,如图7所示:对照组小鼠全部死亡,而实验组有30%的小鼠存活,并且对照组的小鼠平均发病死亡时间比实验组短。说明在活体水平,感染狂犬病毒后,过表达先导RNA1可以一直病毒在活体的复制,推迟发病时间,降低死亡率。通过上述实施例2~3的方法步骤,也可以验证先导RNA2~7用于制备治疗和预防狂犬病的药品。其它未详细说明的部分均为现有技术。尽管上述实施例对本发明做出了详尽的描述,但它仅仅是本发明一部分实施例,而不是全部实施例,人们还可以根据本实施例在不经创造性前提下获得其他实施例,这些实施例都属于本发明保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1