(R)‑1‑叔丁氧羰基‑3‑苄基‑3‑甲酸哌啶的合成及手性拆分方法与流程
本发明属于含N杂环医药中间体的合成及手性拆分技术领域,具体涉及一种(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶的合成及手性拆分方法。
背景技术:
(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶是一种重要的医药中间体,是阿拉莫林的重要合成原料。日前,在西班牙马德里举行的2014年欧洲医学肿瘤学学会(ESMO)发布消息称,一种新药阿拉莫林有望成为有史以来第一款可有效改善癌性恶病质的药物。药物阿拉莫林可产生与所谓的“饥饿激素”肽相同的效果而刺激饥饿感的产生,“饥饿激素”可调节食欲。阿拉莫林可增强患者的食欲,也有助于患者保持健康。近年来的研究结果表明,阿拉莫林有助于晚期NSCLC患者增加体重,这一药物有望用于癌性恶病质的治疗。因此,市场上对该类医药中间体的需求日益增大,(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶的结构式如下:
目前,合成(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶的工艺路线主要是以3-哌啶甲酸为原料,经氯化亚砜脂化后,再与二碳酸二叔丁酯发生加成反应,经LDA(二异丙基胺基锂)还原后再次与溴化苄发生加成反应,然后脱Boc与D-酒石酸制成共同体系被拆分得到的R型构体后再次与二碳酸二叔丁酯发生加成反应制得。其中,找寻适用有效的手性选择体、拆分剂是手性药物拆分的关键。此过程冗杂繁琐、产率较低,经过了两次二碳酸二叔丁酯加成反应,并且经拆分后的手性物质,参加后面的反应过程易发生外消旋化。另外,D-酒石酸有一定的拆分效果,但是达不到完全拆分开两个异构体的目的,其拆分效果比较差,需经过多次拆分才能得到纯度和光学性质都比较好的产品。
技术实现要素:
本发明的目的在于克服现有技术存在的上述缺陷,提供一种(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶的合成及手性拆分方法,该方法具有原料成本低、反应易于控制、产率高、合成工艺简单等显著优点,具有较好的应用前景。本发明的技术方案如下:
(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶的合成及手性拆分方法,包括以下步骤:a将3-哌啶甲酸和氯化亚砜(SOCl2)混合,发生脂化反应合成3-哌啶甲酸乙酯;b在碱性条件下,由3-哌啶甲酸乙酯和二碳酸二叔丁酯((Boc)2O)发生加成反应合成1-叔丁氧羰基-3-甲酸乙酯哌啶;c由1-叔丁氧羰基-3-甲酸乙酯哌啶、正丁基锂、二异丙胺和溴化苄反应合成1-叔丁氧羰基-3-苄基-3-甲酸乙酯哌啶;d由1-叔丁氧羰基-3-苄基-3-甲酸乙酯哌啶与LiOH水合物反应合成1-叔丁氧羰基-3-苄基-3-甲酸哌啶;e由1-叔丁氧羰基-3-苄基-3-甲酸哌啶与R-(+)-α-甲基苄胺进行手性拆分得目标产物。
上述方案中,步骤a包括将3-哌啶甲酸加入到乙醇溶剂中,控制温度滴入氯化亚砜回流反应3-5h,TLC反应完全后脱溶,加入同样体积的乙醇溶剂再次脱溶,溶液冷却结晶即得3-哌啶甲酸乙酯,其中3-哌啶甲酸与氯化亚砜的摩尔比为1:1.2-1.5。
上述方案中,步骤b具体包括以下步骤:
b.1在碱性条件下,将摩尔比为1:1.1-1.5的3-哌啶甲酸乙酯和二碳酸二叔丁酯(Boc)2O加入到乙醇中,控制溶液温度在30℃以下,搅拌过夜,继续反应5-7h;
b.2TLC反应完全后,向溶液中加入乙酸乙酯(EA)进行稀释,控制溶液温度在20℃以下,调节溶液pH至6-7;
b.3溶液有机相采用体积比为3:2的饱和NaHCO3:饱和NaCl混合溶液进行萃取,萃取后所得的有机相干燥后脱溶,接着加入适量石油醚(PE)自然晾干即得。
按照上述方案,步骤c具体包括以下步骤:
c.1将摩尔比为1:1-1.1的正丁基锂和二异丙胺加入到四氢呋喃(THF)或二氯甲烷(DCM)溶剂中,控制溶液温度在-60℃以下,搅拌反应一段时间后得二异丙基胺基锂(LDA)溶液;
c.2向二异丙基胺基锂(LDA)溶液中慢慢滴加1-叔丁氧羰基-3-甲酸乙酯哌啶,再次搅拌反应一段时间,其中1-叔丁氧羰基-3-甲酸乙酯哌啶与正丁基锂的摩尔比为1:1;
c.3反应完成后向溶液中缓慢滴加溴化苄,反应一段时间后将溶液缓慢升至室温,搅拌反应过夜,其中溴化苄与正丁基锂的摩尔比为1:1;
c.4TLC反应完全后,向溶液中加入饱和NH4Cl溶液淬灭,接着用乙酸乙酯(EA)进行萃取,TLC检测水相无产物后再次用二氯甲烷(DCM)萃取。
c.5合并有机相并干燥,接着脱溶,再用体积比为1:10的乙酸乙酯:石油醚混合溶液重结晶即得。
按照上述方案,步骤d包括将摩尔比为1:4的1-叔丁氧羰基-3-苄基-3-甲酸乙酯哌啶和LiOH·H2O加入到体积分数为50%的乙醇水溶液中,升温至105℃回流反应5-7h,TLC反应完全后冷却,用乙酸乙酯萃取,保持溶液温度在20℃以下,调节溶液pH至3,过滤即得。
按照上述方案,步骤e包括将摩尔比为1:6的1-叔丁氧羰基-3-苄基-3-甲酸哌啶和R-(+)-α-甲基苄胺加入到乙酸乙酯溶液中反应,TLC反应完全后过滤,用NaHSO4:乙酸乙酯混合溶液溶解沉淀,水层用乙酸乙酯萃取,合并后的有机相用NaCl溶液洗涤,经干燥、脱溶、重结晶即得目标产物。
上述方案中,步骤e中NaHSO4:乙酸乙酯混合溶液由10wt%的NaHSO4溶液与乙酸乙酯按照1:2的体积比配制而成,采用体积比为1:10的乙酸乙酯:正己烷混合溶液对目标产物进行重结晶。
上述方案中,所述碱性条件是通过向反应体系中加入Na2CO3或NaHCO3或NaOH实现的,Na2CO3或NaHCO3或NaOH的加入量为3-哌啶甲酸乙酯摩尔量的3倍。
上述方案中,1-叔丁氧羰基-3-苄基-3-甲酸哌啶与R-(+)-α-甲基苄胺反应的催化剂为三乙胺(Et3N),其加入量为1-叔丁氧羰基-3-苄基3-甲酸哌啶摩尔量的0.5-1.2%。
上述方案中,步骤b.3、c.5和e中所述干燥是指利用无水Na2SO4或无水MgSO4吸附中间产物中的水分。
本发明提供的(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶合成、手性拆分方法具有以下有益效果:
(1)精简了反应历程,缩短了反应时间。同原有合成路径相比,采用本发明方法合成同样质量的医药中间体,所需时间为原来的一半。
(2)发现了一种新的可替代的水解物质LiOH·H2O,简化了反应流程,提高了反应产率。
(3)工艺简单,操作方便,适合工业化生产。
附图说明
图1为本发明实施例1第一步反应合成的3-哌啶甲酸乙酯核磁谱图;
图2为本发明实施例1第二步反应合成的1-叔丁氧羰基-3-甲酸乙酯哌啶核磁谱图;
图3为本发明实施例1第三步反应合成的1-叔丁氧羰基-3-苄基-3-甲酸乙酯哌啶核磁谱图;
图4为本发明实施例1第四步反应合成的1-叔丁氧羰基-3-苄基-3-甲酸哌啶核磁谱图;
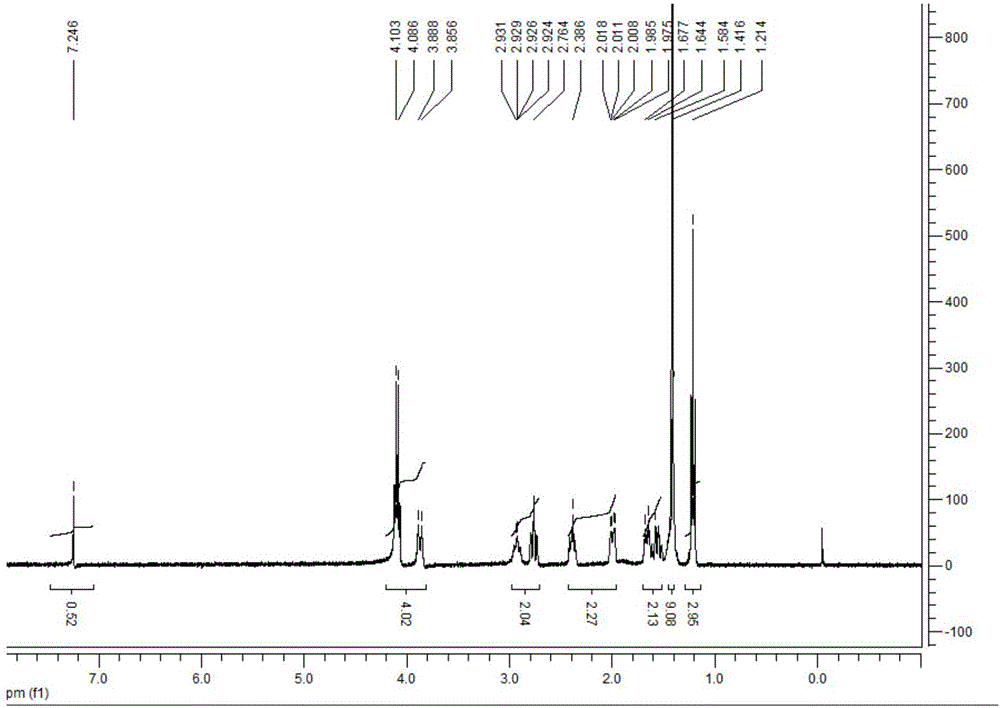
图5为本发明实施例1第五步手性拆分(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶的核磁谱图;
图6为本发明合成及手性拆分路线示意图。
具体实施方式
为使本领域普通技术人员充分理解本发明的技术方案和有益效果,以下结合具体实施例及附图进行进一步充分说明。
本发明提供的医药中间体(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶合成及手性拆分方法以3-哌啶甲酸为原料,通过酯化反应、加成反应、LDA拔氢还原反应、加成反应、手性拆分等五个步骤,方便地合成了目的化合物产物。其基本合成路线如图6所示。
实施例1
一种(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶的合成及手性拆分方法,包括以下五个步骤:
1.合成3-哌啶甲酸乙酯。
向500ml三口烧瓶中加入51.6g(0.4mol)3-哌啶甲酸和205ml乙醇,控制温度向其中缓慢滴入71.4g(0.6mol)氯化亚砜,待温度不变后回流反应3~5h。TLC反应完全,脱溶,加入205ml乙醇再次脱溶。最后将所得有机相置于室温下冷却结晶得3-哌啶甲酸乙酯白色固体71.8g,计算产率为92.78%。本步骤制得的产物核磁谱图如图1所示,从谱图中可以得到以下信息:H1NMR 1(400MHz,CDCl3)δ:1.323(s,2H,CH2),1.581(s,3H,CH3),2.471(s,1H,NH),2.924(s,4H,CH2CHCOO CH2),3.119(s,2H,CH2),3.511(s,2H,CH2N)9.242(m,3H,H2O·HCl)。
2.合成1-叔丁氧羰基-3-甲酸乙酯哌啶。
向2L三口烧瓶中加入134.8g(0.94mol)3-哌啶甲酸乙酯、298.9g Na2CO3(2.82mol)和600ml乙醇,搅拌使其溶解并混合均匀。控制溶液温度在30℃以下,向其中慢慢滴加226.1g(1.03mol)的(Boc)2O,室温下搅拌过夜,接着继续反应5~7h。TLC反应完全,向溶液中加入650mlEA进行稀释。将烧瓶置于冰盐浴中控制溶液温度在20℃以下,向溶液中加入适量5wt%的HCl调节pH至6~7后,有机相用体积比为3:2的饱和NaHCO3:NaCl混合溶液萃取两次,得到的有机相用无水Na2SO4干燥,然后脱溶,向其中加入15~20mlPE,自然晾干,得1-叔丁氧羰基-3-甲酸乙酯哌啶粗品240g,产率为95%。本步骤制得的产物核磁谱图如图2所示,从谱图中可以得到以下信息:H1NMR 2(400MHz,CDCl3)δ:1.153(s,3H,CH3),1.517(m,9H,3*CH3),1.781(t,2H,CH2CH3),2.294(t,2H,CH2N),2.917(t,2H,CH2CH2N),4.017(t,4H,CH2C CO CH2)7.331(s,1H,CH2CH CO CH2)。
3.合成1-甲酸叔丁酯基-3-苄基-3-甲酸乙酯哌啶。
向500ml三口烧瓶中加入27.4g(0.28mol)二异丙胺、16g(0.25mol)正丁基锂、160ml THF,搅拌使其溶解并混合均匀。采用干冰乙醇控制溶液的温度在-60℃以下(保持碳负离子的活性,温度过高,碳负离子会有迁移,得不到想要的化合物),搅拌反应30min后,慢慢滴入64g(0.25mol)1-叔丁氧羰基-3-甲酸乙酯哌啶,再次搅拌反应30min后,慢慢滴加42.75g(0.25mol)溴化苄,反应30min后缓慢升至室温,继续搅拌反应过夜。TLC反应完全,向溶液中加入180ml饱和NH4Cl溶液淬灭,接着向烧瓶中加入200ml EA进行萃取。TLC检测水相无产物后用DCM再萃取一次,合并有机相,用无水Na2SO4干燥,然后浓缩至半干。按照体积比PE:EA=10:1的比例向所得浓缩物中加入PE:EA混合溶液25ml进行重结晶,得白色固体72.52g,产率为83.6%。本步骤制得的产物核磁谱图如图3所示,从谱图中可以得到以下信息:H1NMR 3(400MHz,CDCl3)δ:1.132(s,3H,CH3),1.415(s,9H,3*CH3),1.617(s,2H,CH2),1.624(s,1H,CH),2.109(s,1H,CH CH),2.69(m,1H,CH CH CH),2.814(m,1H,CH CH),3.194(s,2H,CH2),3.619(s,1H,CH),4.194(s,2H,CH2),4.272(s,2H,CH2),7.194(s,2H,CH2),7.219(s,2H,CH2)。
4.合成1-甲酸叔丁酯基-3-苄基-3-甲酸哌啶。
向500ml三口瓶中加入30g(0.086mol)1-叔丁氧羰基-3-苄基3-甲酸乙酯哌啶、14.52g(0.346mol)LiOH·H2O、120mlEtOH和120mlH20,搅拌使其溶解并混合均匀。将溶液升温至105℃回流反应5~7h,TLC反应完全后冷却,加入30mlEA萃取两次,采用冰盐浴控制溶液温度在20℃以下,向其中加入HCl调节pH至3,过滤得白色固体22.68g,产率为82.2%。本步骤制得的产物核磁谱图如图4所示,从谱图中可以得到以下信息:H1NMR 4(400MHz,CDCl3)δ:1.375(s,9H,3*CH3),1.387(m,2H,CH2Ph),1.517(s,1H,CH),1.792(s,1H,CH CH),2.761(m,1H,CH CH),2.811(m,1H,CH CH)3.142(s,2H,NCH2),3.719(m,1H,CCH CH),3.961(s,2H,CHCHC),7.194(s,2H,CH2),7.204(s,1H,COOH),7.219(s,2H,CCH2)。
5.手性拆分(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶。
向500ml三口瓶中加入5g(0.016mol)1-叔丁氧羰基-3-苄基-3-甲酸哌啶、198mlEA、2ml(0.094mol)R-(+)-α-甲基苄胺、0.01gEt3N(0.00008mol)和2mlH20,室温下搅拌反应过夜。TLC反应完全后过滤,过滤所得沉淀用体积比为1:2的NaHSO4溶液(质量分数为10%)与EA的混合溶液溶解。水层用3mlEA萃取,合并得到的有机相用盐水洗涤,接着用无水Na2SO4干燥,真空下脱溶,之后用3ml体积比为1:10的EA:正己烷溶液重结晶,得目标产物白色晶状固体1.9g,产率为76%,终产物的总收率为46%。合成的最终产品为(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶,其核磁谱图如图5所示,从谱图中可以得到以下信息:H1NMR 5(400MHz,CDCl3)δ:1.073(s,9H,3*CH3),1.271(m,1H,CH),1.387(s,1H,CH),1.927(s,2H,CH2),2.181(m,1H,CH CH),2.357(m,1H,CH CH),2.561(m,2H,CH2),2.781(m,2H,CH2),3.176(m,1H,CH),3.248(m,2H,CH2),6.149(m,1H,OH),6.373(s,2H,NH CH2C)。
实施例2
一种(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶的合成及手性拆分方法,包括以下五个步骤:
1.合成3-哌啶甲酸乙酯。
向30L反应釜中加入929g(7.2mol)3-哌啶甲酸和3.7L乙醇,控制温度向其中缓慢滴入1274.4g(10.8mol)氯化亚砜,待温度不变后回流反应3~5h。TLC反应完全,脱溶,向其中加入3.7L乙醇再次脱溶。最后将所得浓缩物置于室温下冷却结晶得3-哌啶甲酸乙酯白色固体1272g,产率91.33%。
2.合成1-叔丁氧羰基-3-甲酸乙酯哌啶。
向30L反应釜中加入1270g(6.56mol)3-哌啶甲酸乙酯、2086g Na2CO3(19.68mol)和5715ml乙醇,搅拌使其溶解并混合均匀。控制溶液温度在30℃以下,向其中慢慢滴加1574g(7.22mol)的(Boc)2O,室温下搅拌过夜,接着继续反应5~7h。TLC反应完全,向溶液中加入适量EA进行稀释。将烧瓶置于冰盐浴中控制溶液温度在20℃以下,向溶液中加入适量5wt%的HCl调节pH至6~7。然后有机相用体积比为3:2的饱和NaHCO3:NaCl混合溶液萃取两次,得到的有机相用无水Na2SO4干燥,脱溶,向其中加入180-250ml PE,自然晾干,得1-叔丁氧羰基-3-甲酸乙酯哌啶粗品1428g,产率为84.66%。
3.合成1-甲酸叔丁酯基-3-苄基-3-甲酸乙酯哌啶。
向30L反应釜中加入471.6g(4.67mol)二异丙胺、299g(4.67mol)正丁基锂、7.2L THF,搅拌使其溶解并混合均匀。采用干冰乙醇控制溶液的温度在-60℃以下,搅拌反应30min后,慢慢滴入1200g(4.67mol)1-叔丁氧羰基-3-甲酸乙酯哌啶,再次搅拌反应30min后,慢慢滴加798.4g(4.67mol)溴化苄,反应30min后缓慢升至室温,搅拌反应过夜。TLC反应完全,向溶液中加入1500ml饱和NH4Cl溶液淬灭,接着向烧瓶中加入适量EA进行萃取。TLC检测水相无产物后用DCM再萃取一次,合并有机相,用无水Na2SO4干燥,接着脱溶,按照PE:EA体积比10:1的的比例向其中加入PE:EA混合溶液220ml进行重结晶,得白色固体1334.4g,产率82.36%。
4.合成1-甲酸叔丁酯基-3-苄基-3-甲酸哌啶。
向30L反应釜中加入900g(2.59mol)1-叔丁氧羰基-3-苄基-3-甲酸乙酯哌啶、435.7g LiOH·H2O(10.37mol)、2.8L EtOH和2.8L H20,搅拌使其溶解并混合均匀。将溶液升温至105℃回流反应5~7h,TLC反应完全后冷却,加入适量EA萃取两次,采用冰盐浴控制溶液温度在20℃以下,向其中加入HCl调节pH至3,过滤得白色固体587.3g,产率70.98%。
5.手性拆分(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶。
向30L反应釜中加入587.3g(1.84mol)1-叔丁氧羰基-3-苄基3-甲酸哌啶、23.18L EA、1292g(11.04mol)R-(+)-α-甲基苄胺、2.23g Et3N(0.022mol)和231.8ml H2O,室温下搅拌反应过夜。TLC反应完全后过滤,过滤所得沉淀用体积比为1:2的10wt%NaHSO4与EA的混合溶液溶解。水层用300mlEA萃取,合并得到的有机相用盐水洗涤,然后用无水Na2SO4干燥,真空下脱溶,之后用100ml体积比为1:10的EA:正己烷溶液重结晶,得最终产物白色晶状固体243.2g,产率82.82%。终产物的总收率为37.44%。
实施例3
一种(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶的合成及手性拆分方法,包括以下五个步骤:
1.合成3-哌啶甲酸乙酯。
在其他参数和条件不变的条件下,参照实施例2的反应步骤,由929g(7.2mol)3-哌啶甲酸和1028g(8.64mol)氯化亚砜反应3-哌啶甲酸乙酯。
2.合成1-叔丁氧羰基-3-甲酸乙酯哌啶。
在其他参数不变和条件的条件下,参照实施例2的反应步骤,由1270g(6.56mol)3-哌啶甲酸乙酯、787g NaOH(19.68mol)和2145g(9.84mol)(Boc)2O反应生成1-叔丁氧羰基-3-甲酸乙酯哌啶。
3.合成1-甲酸叔丁酯基-3-苄基-3-甲酸乙酯哌啶。
在其他参数和条件不变的条件下,参照实施例2的反应步骤,在同样用量的DCM溶剂中合成1-甲酸叔丁酯基-3-苄基-3-甲酸乙酯哌啶。
后续步骤4和步骤5与实施例2相同,最终合成出目标产物(R)-1-叔丁氧羰基-3-苄基-3-甲酸哌啶。
显然,上述实施例仅仅是为清楚地说明所作的实例,而并非对实施方式的限制。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而因此所引申的显而易见的变化或变动仍处于本发明创造的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种(e)-叔丁基-5-(2-硝基乙烯基)-1h-咪唑-1-甲酸叔丁酯及其制备方法
- 一种(r)-2-(n-叔丁氧羰基氨基)联苯丙醇的制备方法
- 一种叔丁基二氧基-3,8-二氮杂二环[3.2.1]辛烷-8-羧酸的制备方法
- 六叔丁基-六己氧基-六苯并[ab,de,lm,op,rs,uv]晕蒄作为MALDI-TOF MS基质在小分子 ...的制作方法
- 一种β-(3,5-二叔丁基-4-羟基苯基)丙酸甲酯含量的测定方法
- 一种三[2.4-二叔丁基苯基]亚磷酸酯釜残的回收方法
- 聚异戊二烯和聚对叔丁基苯乙烯的共混复合阻尼材料的制作方法
- 三叔丁氧基氢化铝锂的制备方法
- 一种GaAs基发光二极管的湿氧氧化方法
- 一类含叔丁基脒结构sglt2/sglt1双靶点抑制剂及用图