卡巴他赛药物前体的制备方法和应用与流程
本发明属于药物化学及制剂领域,具体涉及一系列卡巴他赛药物前体及其制备方法和应用。
背景技术:
卡巴他赛(cabazitaxel,CTX)是一种半合成紫杉烷类化合物(C.J.Paller and E.S.Antonarakis.Cabazitaxel:a novel second-line treatment for metastatic castration-resistant prostate cancer,Drug Des.Dev.Ther.,2011,5,117-124)。其前体从紫杉树针叶中提取获得,化学名称为苯甲酸{(2α,5β,7β,10β,13α)-4-乙酰基-13-{(2R,3S)-3-[(叔丁氧基羰基)氨基]-2-羟基-3-苯丙酰氧基}-1-羟基-7,10-二甲氧基-9-氧代-5,20-环氧紫杉烷-11-烯-2-基}酯,结构式如下:
CTX是一种广谱的抗癌小分子药物,其抑制肿瘤细胞的作用机制跟紫杉醇及多西紫杉醇类似,主要通过促进微管聚合,降低微管的解聚速度,从而使微管处于非功能性状态,达到阻止肿瘤细胞有丝分裂和增殖的目的(C.Villanueva,et al.,Cabazitaxel:A Novel Microtubule Inhibitor,Drugs,2011,71,1251-125)。但是与其他紫杉烷类药物不同的是,CTX与肿瘤的耐药密切相关的P-糖蛋白(P-glycoprotein)的亲和力较低,因此能够克服紫杉醇及多西紫杉醇带来的耐药问题。此外,CTX具有更强的抑制肿瘤细胞增殖的能力,于2010年6月,CTX由美国FDA批准用于激素难治性前列腺癌的治疗。
尽管CTX具有良好的临床应用前景,但是其水溶性差,临床上使用CTX注射液(商品名为JEVTANA,由赛诺菲-安万特公司研发)需要通过添加表面活性剂聚山梨脂80(吐温80)及乙醇作为助溶剂来增加药物的溶解性。但该增溶方法存在诸多缺点:首先吐温80易引发过敏反应,患者用药前需要进行抗过敏治疗;其次,吐温80的血液性毒性较高,成为限制治疗剂量的主要因素;再次药物稳定性差,经稀释后易沉淀析出。此外,临床应用CTX注射液发现具有非常严重的骨髓抑制等副作用,在临床I期的试验结果显示其药物最大耐受剂量(maximum tolerated dose,MTD)仅为25mg/m2,远低于紫杉醇的175mg/m2跟多西紫杉醇的60-100mg/m2的最大耐受剂量(Alain C.Mita,et al.,Phase I and Pharmacokinetic Study of XRP6258(RPR116258A),a Novel Taxane,Administered as a 1-Hour Infusion Every 3Weeks in Patients with Advanced Solid Tumors)。综上,为降低CTX的系统性毒性,需要对该药物的核心分子进行重新设计以提高其临床应用潜力及适应症。
在本发明中我们通过对CTX的羟基位点进行疏水基团后,可在不损伤药物抗肿瘤活性的前提下,通过前药小分子的自组装形成纳米药物的方式或者溶于两亲性表面活性剂或者两亲性高分子聚合物胶束增加CTX的水溶性。此外更为重要的是,我们惊奇地发现,该技术大大提高了MTD,可扩大CTX的临床应用范围和应用前景。
技术实现要素:
为了解决卡巴他赛水溶性差,药物稳定性差,药物体内最大耐受剂量低等问题,本发明提供了一种卡巴他赛(CTX)药物前体,CTX药物前体结构式如式(I)所示:
其中R表示疏水性基团。
本发明中,所述的CTX药物前体由疏水性基团与CTX通过可断裂的酯键连接,利于前体药物在体内通过水解的方式直接释放抗肿瘤成分。
本发明中,所述的疏水性基团由脂肪酸提供。所述脂肪酸可选择饱和脂肪酸或不饱和脂肪酸,优选为不饱和脂肪酸。
作为优选,所述脂肪酸的碳链长度为C2~C22。
更为优选地,所述脂肪酸的碳链长度为C18~C22。
相比于饱和脂肪酸,不饱和脂肪酸修饰的CTX在水中具有更高的溶解度并能够更好地在水中通过自组装的方式形成纳米粒颗粒。所述不饱和脂肪酸优选为DHA(二十二碳六烯酸)、亚麻酸、油酸或亚油酸,更优选为DHA、亚麻酸或亚油酸,所述饱和脂肪酸可选用硬脂酸。
本发明提供的CTX药物前体既能溶解于吐温等两亲性表面活性剂,又可以溶解于水,这大大增加了CTX的应用范围。
同时,本发明还提供了所述CTX药物前体式(1)化合物的制备方法,包括:
(1)在缩合剂存在下,CTX与脂肪酸发生酯化反应;
作为优选,所述缩合剂为N,N’-二环己基碳二亚胺(DCC),1-(3-二甲氨基丙基)-3-乙基碳二亚胺(EDC),N,N’-二异丙基碳二亚胺(DIPC),六氟磷酸苯并三唑-1-基-氧基三吡咯烷基(PyBOP),三吡咯烷基溴化鏻六氟磷酸盐(PyBroP)。
(2)反应完成后对粗产物进行分离纯化,获得结构式如式(Ⅰ)所示的CTX药物前体,分离纯化步骤主要用于除去反应中未反应的原料以及所用的缩合剂、催化剂等杂质。
在第(1)步反应中,反应溶剂选自但不限于二氯甲烷,DMF(二甲基甲酰胺)、DMSO(二甲基亚砜)或者上述溶剂两种或者三种的混合液。
优选的,可以在第(1)步反应中加入碱或碱/催化剂,所述的催化剂选自但不限于DMAP(4-二甲氨基吡啶);所述的碱选自但不限于DIEA(N,N-二异丙基乙胺),Et3N(三乙胺)。
优选的步骤(1)在室温到60℃进行反应。
本发明中所述CTX可以按照姚全兴,李娅,朱小锋,李靖,一种卡巴他赛的合成新方法,化学与生物工程2014年第31卷第2期55-57页中提供的方法制备。
本发明还提供了所述CTX药物前体在制备抗肿瘤药物中的应用。
与现有技术相比,本发明的有益效果为:
(1)本发明的CTX药物前体比临床药物CTX具有相类似的抗肿瘤活性,但是动物体内的耐受剂量大大提高。在体内能够以缓慢水解的方式释放抗癌活性成分,有效减小直接注射CTX造成的体内毒性。
(2)本发明的CTX药物前体在水中具有较好的溶解性(达9mg/mL),避免了制剂辅料的使用,并且具有较高的体内外稳定性。
(3)本发明通过单步酯化法即可获得所述药物前体,产率高,制备成本低,稳定性高,安全性好,符合临床用药的要求,符合大规模工业化生产的要求,具备良好的市场前景与临床应用价值。
附图说明
图1为实施例1中含不饱和烷烃链化合物的CTX前体药物1的合成路线;
图2为实施例2中含不饱和烷烃链化合物的CTX前体药物2的合成路线;
图3为实施例3中含不饱和烷烃链化合物的CTX前体药物3的合成路线;
图4为实施例4中含饱和烷烃链化合物的CTX前体药物4的合成;
图5为实施例5中含饱和烷烃链化合物的CTX前体药物5的合成;
图6为实施例6中含饱和烷烃链化合物的CTX前体药物6的合成;
图7为实施例7中含饱和烷烃链化合物的CTX前体药物7的合成;
图8为实施例1制备的CTX药物前体1在水相中自组装形成纳米粒的透射电镜图;
图9为实施例3制备的CTX药物前体3在水相中自组装形成纳米粒的透射电镜图;
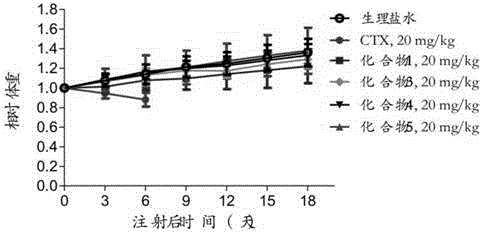
图10-15依次为实施例1、3、4和5中制备的CTX药物前体1,3,4,5在ICR小鼠体内毒性。以生理盐水及CTX(溶于吐温80/乙醇体积1:1)作为对照;
其中,CTX表示卡巴他赛,DHA表示顺式-4,7,10,13,16,19-二十二碳六烯酸,DCM表示二氯甲烷,EDC表示1-(3-二甲氨基丙基)-3-乙基碳二亚胺的盐酸盐,DMAP表示4-二甲氨基吡啶,DIEA表示N,N-二异丙基乙胺。
具体实施方式
下面结合附图和具体实施方式对本发明作进一步详细说明。
实施例1 含不饱和烷烃链化合物的CTX前体药物1的合成
在100mL圆底烧瓶中加入CTX(100mg,0.12mmol)和顺式-4,7,10,13,16,19-二十二碳六烯酸(DHA,39mg,0.12mmol),溶解在5mL无水DCM(二氯甲烷)中,再加入EDC·HCl(25mg,0.13mmol),DMAP(4-二甲氨基吡啶)(16mg,0.13mmol)和DIEA(N,N-二异丙基乙胺)(21μL,0.13mmol)。25℃搅拌过夜,然后分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗;有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂;固体用柱层析色谱分离纯化(DCM:MeOH=200:1)后得到产物1(80mg,产率58%)。
产物1的1H NMR核磁数据以及质谱数据如下:
1H NMR(400MHz,CDCl3):δ0.86-0.90(t,3H),1.20-1.22(d,6H,J=6.4),1.25(s,3H),1.35(s,9H),1.57-1.63(t,2H),1.76-1.82(m,2H),2.00(s,3H),2.04-2.09(t,2H),2.32-2.35(t,3H),2.39-2.49(m,5H),2.77-2.86(m,11H),3.30(s,3H),3.44(s,3H),3.84-3.92(m,2H),4.16-4.18(d,1H,J=8.8),4.30-4.33(d,1H,J=8.4),4.83(s,1H),4.99-5.01(d,1H,J=4.8),5.34-5.37(m,12H),5.64-5.66(d,1H,J=7.2),6.24-6.29(m,1H),7.29-7.31(t,3H),7.38-7.41(t,2H),7.48-7.52(t,2H),7.59-7.63(t,1H),8.10-8.12(d,2H,J=7.2).
HR-ESI Qq-LTMS:计算值[C67H87NO15]+[M+H]+=1146.6148;检测值:1146.6171.
实施例2 含不饱和烷烃链化合物的CTX前体药物2的合成
在100mL圆底烧瓶中加入CTX(100mg,0.12mmol)和亚麻酸(34mg,0.12mmol),溶解在5mL无水二氯甲烷中,再加入EDC·HCl(25mg,0.13mmol),4-二甲氨基吡啶(16mg,0.13mmol)和N,N-二异丙基乙胺(21μL,0.13mmol)。25℃搅拌过夜,然后分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗;有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂;固体用柱层析色谱分离纯化(DCM:MeOH=200:1)后得到产物2(115mg,产率87%)。
产物2的1H NMR核磁数据以及质谱数据如下:
1H NMR(400MHz,CDCl3):δ0.86-0.90(t,3H),1.22(s,9H),1.26(s,8H),1.35(s,9H),1.51-1.55(t,2H),1.62-1.67(m,2H),1.76-1.82(m,2H),2.00(s,3H),2.04-2.08(m,4H),2.31-2.37(m,3H),2.45(s,3H),2.67-2.74(m,1H),2.76-2.79(t,4H),3.30(s,3H),3.44(s,3H),3.84-3.92(m,2H),4.16-4.18(d,1H,J=8.8),4.30-4.33(d,1H,J=8.4),4.83(s,1H),4.99-5.01(d,1H,J=4.8),5.34-5.37(m,6H),5.64-5.66(d,1H,J=7.2),6.24-6.29(m,1H),7.29-7.31(t,3H),7.38-7.41(t,2H),7.48-7.52(t,2H),7.59-7.63(t,1H),8.10-8.12(d,2H,J=7.2).
HR-ESI Qq-LTMS:计算值[C63H85NO15]+[M+H]+=1096.5992;检测值:1096.6005.
实施例3 含不饱和烷烃链化合物的CTX前体药物3的合成
在100mL圆底烧瓶中加入CTX(100mg,0.12mmol)和亚油酸(34mg,0.12mmol),溶解在5mL无水二氯甲烷中,再加入EDC·HCl(25mg,0.13mmol),4-二甲氨基吡啶(16mg,0.13mmol)和N,N-二异丙基乙胺(21μL,0.13mmol)。25℃搅拌过夜,然后分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗;有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂;固体用柱层析色谱分离纯化(DCM:MeOH=200:1)后得到产物3(120mg,产率91%)。
产物3的1H NMR核磁数据以及质谱数据如下:
1H NMR(400MHz,CDCl3):δ0.86-0.90(t,3H),1.22(s,9H),1.26(s,14H),1.35(s,9H),1.51-1.55(t,2H),1.62-1.67(m,2H),1.76-1.82(m,2H),2.00(s,3H),2.04-2.08(m,4H),2.31-2.37(m,3H),2.45(s,3H),2.67-2.74(m,1H),2.76-2.79(t,2H),3.30(s,3H),3.44(s,3H),3.84-3.92(m,2H),4.16-4.18(d,1H,J=8.8),4.30-4.33(d,1H,J=8.4),4.83(s,1H),4.99-5.01(d,1H,J=4.8),5.34-5.37(m,4H),5.64-5.66(d,1H,J=7.2),6.24-6.29(m,1H),7.29-7.31(t,3H),7.38-7.41(t,2H),7.48-7.52(t,2H),7.59-7.63(t,1H),8.10-8.12(d,2H,J=7.2).
HR-ESI Qq-LTMS:计算值[C63H87NO15]+[M+H]+=1098.6148;检测值:1098.6166.
实施例4 含不饱和烷烃链化合物的CTX前体药物4的合成
在100mL圆底烧瓶中加入CTX(100mg,0.12mmol)和油酸(34mg,0.12mmol),溶解在5mL无水DCM(二氯甲烷)中,再加入EDC·HCl(25mg,0.13mmol),DMAP(4-二甲氨基吡啶)(16mg,0.13mmol)和DIEA(N,N-二异丙基乙胺)(21μL,0.13mmol)。25℃搅拌过夜,然后分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗;有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂;固体用柱层析色谱分离纯化(DCM:MeOH=200:1)后得到产物4(98mg,产率76%)。
产物4的1H NMR核磁数据以及质谱数据如下:
1H NMR(400MHz,CDCl3):δ0.86-0.90(t,3H),1.22(s,9H),1.26(s,20H),1.35(s,9H),1.51-1.55(t,2H),1.62-1.67(m,2H),1.76-1.82(m,2H),2.00(s,7H),2.31-2.37(m,3H),2.45(s,3H),2.67-2.74(m,1H),3.30(s,3H),3.44(s,3H),3.84-3.92(m,2H),4.16-4.18(d,1H,J=8.8),4.30-4.33(d,1H,J=8.4),4.83(s,1H),4.99-5.01(d,1H,J=4.8),5.34-5.37(m,2H),5.64-5.66(d,1H,J=7.2),6.24-6.29(m,1H),7.29-7.31(t,3H),7.38-7.41(t,2H),7.48-7.52(t,2H),7.59-7.63(t,1H),8.10-8.12(d,2H,J=7.2).
HR-ESI Qq-LTMS:计算值[C63H89NO15]+[M+H]+=1100.6305;检测值:1100.6265.
实施例5 含饱和烷烃链化合物的CTX前体药物5的合成
在100mL圆底烧瓶中加入CTX(CTX)(100mg,0.12mmol)和硬脂酸(34mg,0.12mmol),溶解在5mL无水DCM(二氯甲烷)中,再加入EDC·HCl(25mg,0.13mmol),DMAP(4-二甲氨基吡啶)(16mg,0.13mmol)和DIEA(N,N-二异丙基乙胺)(21μL,0.13mmol)。25℃搅拌过夜,然后分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗;有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂;固体用柱层析色谱分离纯化(DCM:MeOH=200:1)后得到产物5(93mg,产率70%)。
产物5的1H NMR核磁数据以及质谱数据如下:
1H NMR(400MHz,CDCl3):δ0.86-0.90(t,3H),1.22(s,9H),1.26(s,28H),1.35(s,9H),1.51-1.55(t,2H),1.62-1.67(m,2H),1.76-1.82(m,2H),2.00(s,3H),2.31-2.37(m,3H),2.45(s,3H),2.67-2.74(m,1H),3.30(s,3H),3.44(s,3H),3.84-3.92(m,2H),4.16-4.18(d,1H,J=8.8),4.30-4.33(d,1H,J=8.4),4.83(s,1H),4.99-5.01(d,1H,J=4.8),5.64-5.66(d,1H,J=7.2),6.24-6.29(m,1H),7.29-7.31(t,3H),7.38-7.41(t,2H),7.48-7.52(t,2H),7.59-7.63(t,1H),8.10-8.12(d,2H,J=7.2).
HR-ESI Qq-LTMS:计算值[C63H91NO15]+[M+H]+=1102.6461;检测值:1102.6479.
实施例6 含饱和烷烃链化合物的CTX前体药物6的合成
在100mL圆底烧瓶中加入CTX(CTX)(100mg,0.12mmol)和庚酸(16mg,0.12mmol),溶解在5mL无水DCM(二氯甲烷)中,再加入EDC·HCl(25mg,0.13mmol),DMAP(4-二甲氨基吡啶)(16mg,0.13mmol)和DIEA(N,N-二异丙基乙胺)(21μL,0.13mmol)。25℃搅拌过夜,然后分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗;有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂;固体用柱层析色谱分离纯化(DCM:MeOH=200:1)后得到产物6(80mg,产率71%)。
产物6的1H NMR核磁数据以及质谱数据如下:
1H NMR(400MHz,CDCl3):δ0.86-0.90(t,3H),1.22(s,9H),1.26(s,6H),1.35(s,9H),1.51-1.55(t,2H),1.62-1.67(m,2H),1.76-1.82(m,2H),2.00(s,3H),2.31-2.37(m,3H),2.45(s,3H),2.67-2.74(m,1H),3.30(s,3H),3.44(s,3H),3.84-3.92(m,2H),4.16-4.18(d,1H,J=8.8),4.30-4.33(d,1H,J=8.4),4.83(s,1H),4.99-5.01(d,1H,J=4.8),5.64-5.66(d,1H,J=7.2),6.24-6.29(m,1H),7.29-7.31(t,3H),7.38-7.41(t,2H),7.48-7.52(t,2H),7.59-7.63(t,1H),8.10-8.12(d,2H,J=7.2).
HR-ESI Qq-LTMS:计算值[C52H69NO15]+[M+H]+=948.4740;检测值:948.4753.
实施例7 含饱和烷烃链化合物的CTX前体药物7的合成
在100mL圆底烧瓶中加入CTX(CTX)(100mg,0.12mmol)和乙酸(7.2mg,0.12mmol),溶解在5mL无水DCM(二氯甲烷)中,再加入EDC·HCl(25mg,0.13mmol),DMAP(4-二甲氨基吡啶)(16mg,0.13mmol)和DIEA(N,N-二异丙基乙胺)(21μL,0.13mmol)。25℃搅拌过夜,然后分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗;有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂;固体用柱层析色谱分离纯化(DCM:MeOH=200:1)后得到产物7(95mg,产率90%)。
产物7的1H NMR核磁数据以及质谱数据如下:
1H NMR(400MHz,CDCl3):δ1.22(s,9H),1.35(s,9H),1.51-1.55(t,2H),1.76-1.82(m,2H),2.00(s,3H),2.31-2.37(m,3H),2.42(s,3H),2.45(s,3H),2.67-2.74(m,1H),3.32(s,3H),3.46(s,3H),3.84-3.92(m,2H),4.16-4.18(d,1H,J=8.8),4.31-4.34(d,1H,J=8.4),4.85(s,1H),5.00-5.02(d,1H,J=4.8),5.68-5.71(d,1H,J=7.2),6.26-6.30(m,1H),7.20-7.31(t,3H),7.40-7.43(t,2H),7.49-7.53(t,2H),7.58-7.59(t,1H),8.11-8.13(d,2H,J=7.2).
HR-ESI Qq-LTMS:calcd for[C47H60NO15]+[M+H]+=878.3957;obsd 878.4012.
实施例8 CTX前药在水相中的自组装能力测试
取实施例1、3中制备得到的CTX前药1与3各10mg,溶于100μl的二甲基亚砜(DMSO)中,再缓慢注入水中,形成澄清的溶液;经水溶液中透析后即可除去DMSO溶液。上述水溶液使用透射电镜(TEM)观察颗粒的形貌,结果如图8和9。
实施例9 CTX前药抑制肿瘤细胞增殖的能力
考察实施例中CTX前药对肿瘤细胞增殖的抑制作用,具体方法如下:
取对数生长期细胞,接种于96孔培养板(5000个细胞/孔)。放入在37℃细胞培养箱中恒温培养24h后,加入各纳米药物,取7个浓度梯度,以CTX(溶于二甲亚砜)作为对照组,每种药每个浓度4个重复值,加完药后将96孔细胞板放入细胞培养箱中培养72h后,在96孔板的每孔内加入30μL的四甲基偶氮唑蓝(MTT),继续放入细胞培养箱中培养4h后,吸弃培养基,每孔加入100μL二甲亚砜,用酶标仪检测490nm处的吸光值。计算细胞存活率,得到药物对细胞生长的IC50(半数抑制浓度)。合成的CTX前药对肿瘤细胞的体外毒性结果见表1。
表1.CTX化合物在肿瘤细胞中的MTT活性测试(IC50±SD,μM)。
表1结果显示,与人乳腺癌细胞Bcap-37及人肠癌细胞LoVo共培养72h后,CTX前药4,5与上述细胞共培养72h后,其抑制肿瘤细胞的能力降低较明显。但是CTX前药1-3能够高效诱导肿瘤细胞的凋亡,其IC50值与CTX药物接近。因此细胞毒性实验表明,CTX前药1-3具有较强的抑制肿瘤细胞增殖的能力,具有广阔的抗肿瘤应用前景。
实施例10 药物体内毒性评价实验
取实施例1、3、4、5制备的CTX前药各100mg,加入DSPE-PEG2000(10mg)溶于728μl二甲基亚砜(DMSO)中,再缓慢注入水中,即可形成纳米粒。
ICR小鼠(4-5周龄),每组10只,注射由实施例1、3、4、5制备的CTX药物前体形成的纳米粒,考察尾静脉注射CTX与CTX前药纳米粒(注射三次,每隔两天注射一次)后对小鼠体重的影响,评价药物的体内毒性。实验结果如图10-15所示,小鼠注射CTX前药纳米药物后,体重与空白对照组(未用任何药物组)小鼠体重基本一致,呈上升趋势,即使是高剂量(70mg/kg)用药组小鼠的体重也基本保持稳定,没有出现明显的药物毒性。但注射低剂量的CTX药物时,小鼠的体重呈明显下降趋势;其中剂量为20mg/kg时,小鼠6天后开始死亡。上述实验结果表明,制备的CTX药物前体能够显著降低CTX药物的毒性,因此有望大幅提高CTX药物的临床应用范围,具有非常大的临床应用前景。
- 还没有人留言评论。精彩留言会获得点赞!