用于选择性分离纯化环糊精及其衍生物的光响应分子印迹材料的制备方法与流程
本发明涉及化学与材料和分子分离技术领域,尤其是涉及一种用于选择性分离纯化环糊精及其衍生物的光响应分子印迹材料的制备新方法。
背景技术:
环糊精(Cyclodextrin,简称CD)是由芽孢杆菌属的某些种产生的葡萄糖基转移酶(CGTase)作用于淀粉而生成的一类环状低聚糖。根据葡萄糖单元数目的不同,常见的环糊精分别包含6、7和8个葡萄糖单元(α-CD、β-CD、γ-CD)。环糊精因其特殊的外侧亲水而内腔疏水这一独特的两亲性空腔结构,可作为“主体”分子包结络合不同的“客体”化合物,形成主客体包结络合物。
偶氮苯及其衍生物是一类能发生光致异构的化合物。反式构象是偶氮苯的稳定构象,并能与环糊精进行包结络合。当用紫外光(365nm)照射后,偶氮苯分子由反式构象变为顺式构象。顺式构象偶氮苯不能继续与环糊精进行包结络合作用,而从环糊精空腔内脱落出来。偶氮苯与环糊精之间可逆的包结络合作用为功能性超分子自组装以及光敏材料等领域提供了应用价值。
分子印迹技术(Molecular Imprinting Technology,MIT)是高分子化学、材料化学和生物化学的交叉学科,至今已有70年的发展历史,在色谱分离、固相萃取、生物传感器、模拟酶催化、生物医学等方面得到了日益广泛的研究和开发,展示了广阔的应用前景。利用MIT可以在聚合物中留下与模板分子大小、形状以及功能基团空间排列相匹配的空穴,从而获得对目标分子具有特异性识别能力和高亲和力的分子印迹聚合物(Molecularly Imprinted Polymers,MIPs)。
大多数酶作用于淀粉或其水解物时会转化生成α-CD、β-CD、γ-CD和分支环糊精等,酶转化产物中环糊精及其衍生物的分离纯化要经过脱色、除盐、浓缩等工艺,存在操作复杂,耗时长,收率较低等缺点;若采用纳滤膜,色谱柱等进行分离纯化,其工业化产量不高,且价格昂贵。传统的分离纯化工艺限制了环糊精及其衍生物的工业化生产及其进一步应用。
技术实现要素:
针对现有技术存在的上述问题,一种用于选择性分离纯化环糊精及其衍生物的光响应分子印迹材料的制备方法。本发明制备工艺简单,制备的分子印迹材料化学稳定性好、吸附容量大、重复利用率高,能够在复杂环境中分离纯化环糊精及其衍生物。
本发明的技术方案如下:
一种用于选择性分离纯化环糊精及其衍生物的光响应分子印迹材料的制备方法,包括制备4-羟基偶氮苯的步骤和制备功能单体4-甲基丙烯酰氧偶氮苯的步骤,具体制备方法如下:
(1)于三口烧瓶中依次加入苯胺15.0g、水10~160mL和浓盐酸10~50mL,混合搅拌10~30分钟后,向上述溶液中滴加溶解在20~80mL水中的12.0~14.0g NaNO2溶液;所述浓盐酸的浓度为10~12mol/L;
加料结束后,在冰浴条件下继续搅拌20~60分钟;最后,滴加溶于40~150mL浓度为8~12%的NaOH溶液的苯酚溶液;所述苯酚的用量与苯胺的摩尔量相同;滴加结束后,保持冰浴并继续搅拌40~600分钟;
反应结束后,抽滤,滤饼用水洗涤,再用体积比为1:1的乙醇和水重结晶,得到深橘红色产物4-羟基偶氮苯;
(2)将0.15~1.5g的N,N-二甲基氨吡啶、2.0~40.0g的三乙胺和2.0~10.0g的4-羟基偶氮苯溶解在100.0~200.0mL乙腈中,混合液置于冰浴中;
向混合液中缓慢滴加2.0~20.0g甲基丙烯酰氯和5.0~20.0mL乙腈混合液,滴加结束后,将混合液移至35~60℃的油浴中反应12~48h,冷却至室温,加入30.0~80.0mL饱和食盐水,析出黄色沉淀物,过滤,用浓度为1.5~2.5mol/L的HCl溶液洗涤滤饼,自然晾干后用冰醋酸重结晶,得到功能单体4-甲基丙烯酰氧偶氮苯;
(3)将步骤(2)中得到的功能单体和模板分子混合均匀后加入70~80mL致孔剂,黑暗室温条件下搅拌2~4h;向其中加入交联剂和0.0060g~0.0100g引发剂,冰水浴冷却条件下向澄清溶液中通氮气除氧后密封反应体系,在50~85℃下反应20~48h,得到分子印迹聚合物;
移除分子印迹聚合物中的模板分子,然后进行真空干燥,即得所述光响应分子印迹材料。
所述模板分子、功能单体、交联剂的摩尔之比为1:(1~8):(5~20)。
所述模板分子为α-环糊精、β-环糊精、γ-环糊精、分支环糊精。
所述交联剂为二甲基丙烯酸乙二醇酯即EGDMA。
所述引发剂为偶氮二异丁腈即AIBN。
所述致孔剂为乙腈、甲醇、氯仿中的任意一种。
其中,4-甲基丙烯酰氧偶氮苯(4-methacryloyloxy azobenzene)结构式如下:
本发明有益的技术效果在于:
本发明基于分子印迹技术,合成了一种新型具有较高吸附能力的光响应印迹聚合物(MIPs),并应用于实际样品中目标分析物的光控吸附与分离。通过引入光敏单体4-甲基丙烯酰氧偶氮苯,使分子印迹材料表现出良好的光致异构化特性。在外部紫外光照射下,模板分子(环糊精及其衍生物)可以容易地从印迹位点中分离。同时,在可见光照射下,该聚合物又重新具有对模板分子(环糊精及其衍生物)特异识别的印迹位点,为环糊精及其衍生物高效快速的吸附分离提供新的技术手段。同时,分子印迹材料抗恶劣环境能力强、稳定性高、使用寿命长、易实现工业化,其应用前景广阔。
本发明制备的光响应分子印迹材料具有优异的光响应特性,对环糊精及其衍生物具有分离效果显著,较好的识别性能,可重复使用等优点。这为选择性分离纯化环糊精及其衍生物提供了一种新方法。具体优势在于:
(1)分子印迹材料对模板分子具有专一识别性的特点,能够在复杂环境中选择性吸附分离模板分子;
(2)引入光敏性功能单体,利用可见光(440nm)和紫外光(365nm)交替照射分子印迹材料,使得该材料具有对目标分子进行选择性的吸附与释放,有效解决模板分子洗脱难的问题。
附图说明
图1为本发明的分子印迹聚合物的合成路线示意图;
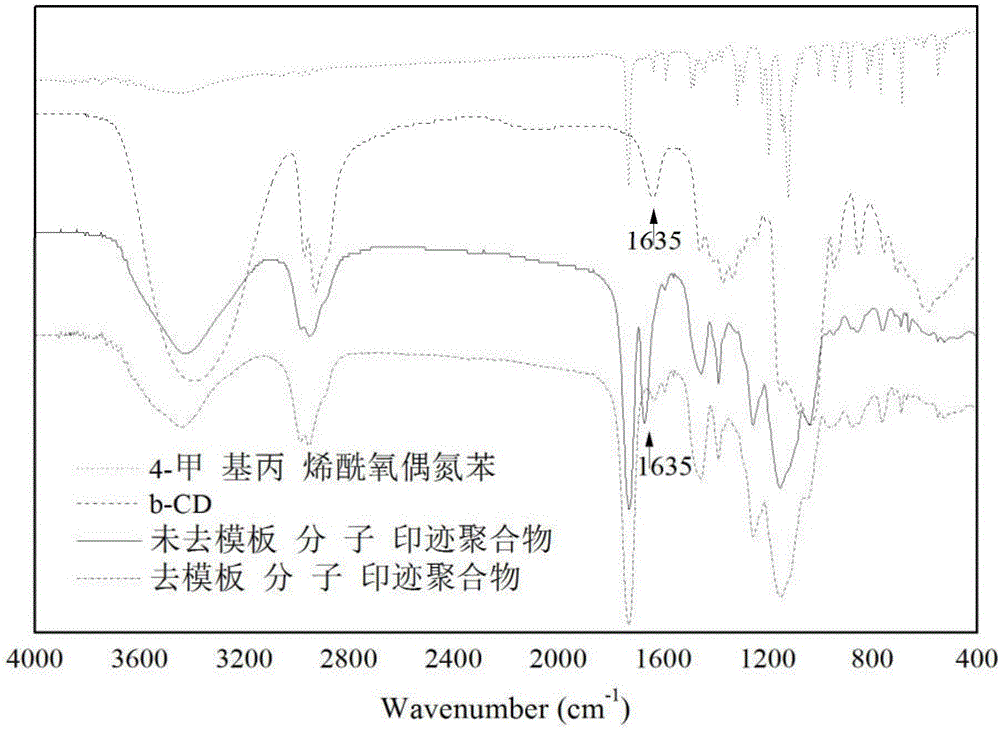
图2为功能单体、模板分子以及合成的分子印迹聚合物去模板前后的红外光谱图;
图3为模板分子以及合成的分子印迹聚合物去模板前后的热重分析图;
图4为功能单体的光致异构紫外-可见图谱,图A为紫外光(365nm)照射下的光致异构紫外-可见图谱,图B为可见光(440nm)照射下的光致异构紫外-可见图谱;
图5为功能单体的光响应性;
图6为分子印迹聚合物对β-CD的吸附等温曲线;
图7为分子印迹聚合物对β-CD的动态吸附曲线;
图8为分子印迹聚合物对α-CD、β-CD、γ-CD和羟丙基环糊精(HP-β-CD)的选择性吸附能力;
图9为分子印迹聚合物对环糊精及其类似物的光控释放和吸收。
具体实施方式
下面结合实施例和附图1,对本发明进行具体描述。
实施例1
(1)苯胺(15g,162mmol),10mL水和10mL浓度为12mol/L的浓盐酸的混合搅拌10min后,滴加溶解在20mL水中的NaNO2(12g)。在冰浴条件下继续搅拌20min后,滴加溶于40mL 8%的NaOH的苯酚溶液。滴加结束后保持冰浴继续搅拌40min。反应结束后,抽滤,滤饼用水洗涤,再用体积比为1:1的乙醇和水的混合液重结晶,得到深橘红色产物4-羟基偶氮苯;
(2)0.15g N,N-二甲基氨吡啶,2.0g三乙胺和2.0g 4-羟基偶氮苯溶解在100.0mL乙腈中,把该混合液置于冰水浴中,缓慢滴加2.0g甲基丙稀酰氯和5.0mL乙腈混合液,移至35℃的油浴中反应48h后,冷却至室温,加入30.0mL饱和食盐水,析出黄色沉淀物,过滤,用1.5mol/L HCl溶液洗涤滤饼,自然晾干。用适量的冰醋酸重结晶得到功能单体4-甲基丙烯酰氧偶氮苯;
(3)将4-甲基丙烯酰氧偶氮苯(0.3990g,1.5mmol)、β-CD(1.7025g,1.5mmol)和70mL无水乙腈依次加入100mL圆底烧瓶中,室温黑暗条件下搅拌2h。向其中加入EGDMA(1.42mL,7.5mmol)和0.0060g AIBN,冰水浴冷却条件下向澄清溶液中通氮气15min除氧,随后将反应瓶封口后置于50℃油浴中反应48h。反应结束后超速离心收集所得聚合物,并用水抽提36h,以洗脱聚合物中残留的模板分子,将所得的分子印迹聚合物进行真空干燥。
实施例2
(1)苯胺(15g,162mmol),80mL水和30mL浓度为11mol/L的浓盐酸的混合搅拌20min后,滴加溶解在30mL水中的NaNO2(13g)。在冰浴条件下继续搅拌40min后,滴加溶于90mL 10%的NaOH的苯酚溶液。滴加结束后保持冰浴继续搅拌300min。反应结束后,抽滤,滤饼用水洗涤,再用体积比为1:1的乙醇和水的混合液重结晶,得到深橘红色产物4-羟基偶氮苯;
(2)0.75g N,N-二甲基氨吡啶,20.0g三乙胺和6.0g 4-羟基偶氮苯溶解在150.0mL乙腈中,把该混合液置于冰水浴中,缓慢滴加10.0g甲基丙稀酰氯和10.0mL乙腈混合液,移至50℃的油浴中反应24h后,冷却至室温,加入50.0mL饱和食盐水,析出黄色沉淀物,过滤,用2mol/L HCl溶液洗涤滤饼,自然晾干。用适量的冰醋酸重结晶得到功能单体4-甲基丙烯酰氧偶氮苯;
(3)将4-甲基丙烯酰氧偶氮苯(1.5960g,6.0mmol)、HP-β-CD(2.1465g,1.5mmol)和75mL无水甲醇依次加入100mL圆底烧瓶中,室温黑暗条件下搅拌3h。向其中加入EGDMA(2.83mL,15.0mmol)和0.0080g AIBN,冰水浴冷却条件下向澄清溶液中通氮气20min除氧,随后将反应瓶封口后置于70℃油浴中反应36h。反应结束后超速离心收集所得聚合物,并用水抽提24h,以洗脱聚合物中残留的模板分子,将所得的分子印迹聚合物进行真空干燥。
实施例3
(1)苯胺(15g,162mmol),160mL水和50mL浓度为10mol/L的浓盐酸的混合搅拌30min后,滴加溶解在40mL水中的NaNO2(14g)。在冰浴条件下继续搅拌60min后,滴加溶于150mL 12%的NaOH的苯酚溶液。滴加结束后保持冰浴继续搅拌600min。反应结束后,抽滤,滤饼用水洗涤,再用体积比为1:1的乙醇和水的混合液重结晶,得到深橘红色产物4-羟基偶氮苯;
(2)1.5g N,N-二甲基氨吡啶,40.0g三乙胺和10.0g 4-羟基偶氮苯溶解在200.0mL乙腈中,把该混合液置于冰水浴中,缓慢滴加20.0g甲基丙稀酰氯和20.0mL乙腈混合液,移至60℃的油浴中反应12h后,冷却至室温,加入80.0mL饱和食盐水,析出黄色沉淀物,过滤,用2.5mol/L HCl溶液洗涤滤饼,自然晾干。用适量的冰醋酸重结晶得到功能单体4-甲基丙烯酰氧偶氮苯;
(3)将4-甲基丙烯酰氧偶氮苯(3.1920g,12.0mmol)、G1-β-CD(1.9457g,1.5mmol)和80mL无水氯仿依次加入100mL圆底烧瓶中,室温黑暗条件下搅拌4h。向其中加入EGDMA(5.68mL,30.0mmol)和0.01g AIBN,冰水浴冷却条件下向澄清溶液中通氮气30min除氧,随后将反应瓶封口后置于85℃油浴中反应20h。反应结束后超速离心收集所得聚合物,并用水抽提48h,以洗脱聚合物中残留的模板分子,将所得的分子印迹聚合物进行真空干燥。
图2为上述实例1功能单体、模板分子以及合成的分子印迹聚合物去模板前后的红外光谱图。从图中可以看出,分子印迹聚合物去模板前后的红外光谱特征峰几乎是重合的。1635cm-1是模板分子β-CD的特征吸收峰,去模板之前的分子印迹聚合物的红外图谱中出现这一特征峰,去模板之后的分子印迹聚合物的红外图谱中这一特征峰消失,说明模板分子β-CD已被去除。
图3为上述实例2模板分子以及合成的分子印迹聚合物去模板前后的热重分析图。从TGA图中可知,去模板之后的分子印迹聚合物的失重主要发生在300-450℃之间,300℃之前和450℃以后继续升温样品重量基本恒定,说明合成的分子印迹聚合物具有良好的热稳定性。对于β-CD和去模板之前的分子印迹聚合物而言,从室温上升到100℃时发生第一次失重,质量损失分别为6.24%和6.29%左右,主要来源于聚合物中水分的蒸发和部分化合物的分解;在300-450℃之间发生第二次失重,质量损失分别为93.08%和91.88%左右,主要源于有机化合物的分解,最后的残留物质主要是碳化残留物。由DTG曲线可知,β-CD和去模板之前的分子印迹聚合物都呈现两次物质损失,在100℃和350℃左右,去模板之后的分子印迹聚合物呈现出一次物质损失,在350℃左右。从图中我们可以得知,与去模板之后的分子印迹聚合物相比,去模板之前的分子印迹聚合物的热稳定性较差,可能与结合的β-CD有关。
图4为实施例3用365nm单色光照射浓度为1.0×10-5mol/L的4-甲基丙烯酰氧偶氮苯紫外吸收光谱随时间的变化情况(图A),在326nm和440nm处有两个特征吸收峰,分别归属为偶氮苯基团的π-π*和n-π*跃迁。随着365nm光照时间的增加,偶氮苯基团从trans→cis,在π-π*跃迁的吸收峰326nm逐渐减少,当光照射50min时达到光化学稳定态;然后改用440nm光照射(图B),随着光照时间的增加,偶氮苯基团从cis→trans构型,在326nm的吸收峰逐渐增加,10min达到稳定态。
用一级动力学方程对功能单体的光致异构化过程进行拟合,计算出单体从trans→cis和cis→trans的光致异构化速率常数分别是2.54×10-3s-1和11.9×10-3s-1。
图5为实施例3在紫外光和可见光的交替照射下(在每个循环中,365nm紫外光照射时间均为50min,440nm可见光照射时间均为10min),功能单体4-甲基丙烯酰氧偶氮苯最大吸收波长下的吸光度数值可以反复循环出现,进一步表明其具有优异的可逆光致异构化性能。
图6为上述实施例3合成的MIPs对HP-β-CD的等温吸附曲线。从图中可知,随着溶液中HP-β-CD浓度增加,分子印迹聚合物对HP-β-CD的吸附量也随之增加。
图7为上述实施例1合成的MIPs与一定浓度的β-CD结合随时间变化的动力吸附曲线。开始吸附时,β-CD快速地结合到MIPs的空腔中,吸附4h达到平衡,约达68%的结合量。
图8为上述实施例1合成的MIPs对于浓度相同、结构类似的四种化合物的选择性吸附实验。MIPs对β-CD显示出较高的结合容量,对β-CD,HP-β-CD,α-CD,γ-CD的吸附量分别为68.3%、51.1%、24.6%和11.3%。对于HP-β-CD,由于其与β-CD有类似的结构,其吸附能力比α-CD和γ-CD高。该结果说明,MIPs的识别机制是基于模板分子的大小、形状和功能性相互作用的结果。
图9为上述实施例1合成的MIPs对环糊精及其类似物的光控释放和吸收从图中可以看出,当紫外光源开启时,MIPs对模板分子β-CD的吸附量降低,特异性吸附显著减弱,而当紫外光源关闭后,MIPs对模板分子β-CD的吸附量逐渐恢复,特异性吸附也逐渐恢复。365nm光照射后,MIPs对模板分子β-CD的吸附量降低,这可能原因是由于在365nm光照射下,偶氮生色团的光致异构化trans→cis使MIPs内部空腔受体位点发生变化,导致受体几何的变化,致使BPA从材料中释放出来。改用440nm光照射后,MIPs对模板分子β-CD的吸附量逐渐恢复,特异性吸附也逐渐恢复。这个定量释放和吸收β-CD过程,是偶氮发色团在光控过程中受体位点的配置和底物亲和力的可逆性证据。重复365-440nm的光照射三个循环,M1Ps对β-CD的释放和吸收类似于前一个周期。对于其他结构类似物,相同实验条件下,结合能力下降的顺序:HP-β-CD>α-CD>γ-CD。通过研究,观察到MIPs材料的对β-CD的光控释放与吸收效果,实现通过波长来控制MIPs材料对模板分子的控制释放与吸收。
- 还没有人留言评论。精彩留言会获得点赞!
- 用于生产化学品及其衍生物的组合物和方法与制造工艺
- 7‑OH‑8‑[(1‑胺基‑1‑苯基衍生物)‑次甲基]‑黄酮化合物及其制备方法及其应用与制造工艺
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 含D型非天然氨基酸的抗菌肽类似物及其合成与应用的制造方法与工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 一种白杨素氨基酸衍生物的制备方法与制造工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 吡唑衍生物及其作为大麻素受体介体的用途的制造方法与工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 水生态处理中淤泥及衍生物的再利用处理系统的制造方法与工艺
- Schiglautone A衍生物的组合物用于制备抗缺氧药物的制作方法
- Schiglautone A衍生物的组合物用于制备抗红细胞低下性贫血药物的制作方法