发酵乳杆菌快速溯源方法、用于所述方法的联合序列及其构建方法与流程
本发明涉及发酵乳杆菌的快速溯源方法,特别是基于多个看家基因构成的联合序列构建的系统发育树进行溯源的方法。
背景技术:
发酵乳杆菌(Lactobacillus fermentum)属于乳杆菌科中的乳杆菌属,革兰氏阳性兼性厌氧菌,且不产芽孢,是人类肠道中主要的异型发酵乳杆菌。目前,大量报道表明发酵乳杆菌(Lactobacillus fermentum)具有水解胆盐、降胆固醇的功能,多用于豆乳发酵、香肠发酵,可阻止有害菌的生长,且对宿主健康具有重要益生作用。
近年来,越来越多的分子方法用于分析发酵乳杆菌(Lactobacillus fermentum)的遗传多样性研究,如采用荧光原位杂交(FISH)和脉冲电场凝胶电泳(PFGE)方法分析人体生殖道Lactobacillus fermentum分离株的遗传差异,但由于这些技术具有分辨率低以及重现性低等特点,很难识别其分离源。
多序列位点分型方法是近年来随着分子生物学技术的不断进步而发展起来的分子生物学分析方法,它应用测序技术分析多个看家基因的核酸序列,通过菌株核苷酸序列的变异进行多样性比较。一个基因突变后形成不同的核苷酸序列被认定为这个基因的等位基因,每个看家基因不同的等位基因按照发现的先后顺序分配一个等位基因序号,将所有看家基因的每个等位基因组合起来构成等位基因图谱,每个独特的等位基因图谱就对应了一个序列型。通过比较序列型可以发现各个菌株的相关性,即密切相关菌株具有相同的序列型或仅有极个别基因座不同的序列型,而不相关菌株的序列型至少有3个或以上基因座不同。通过该分析以反映菌株之间的进化关系,并对菌株进行分型研究。MLST系统的结合了序列测定的高通量特性和已经建立的群体遗传学技术,能够应用于细菌的群体生物学、流行病学、致病性及生物进化研究。与传统的分子分型方法比较,MLST系统具有简便快速、重复性强分辨率高等优点。
于洁等人研究了中国、蒙古和俄罗斯传统发酵乳制品中嗜热链球菌的MLST分析(中国、俄罗斯和蒙古国地区传统发酵乳制品中嗜热链球菌的多位点序列分型研究,2013.06),其以嗜热链球菌ND03的全基因组序列为基础,分散选取了10个单拷贝的等位基因作为MLST分型研究的靶基因,应用Mega5.0软件,通过聚类分析构建系统发育树,再应用eBURST软件分析世界范围内不同地区发酵乳中嗜热链球菌分离株的菌群结构,实现嗜热链球菌的溯源。
然而因物种不同,以及文献中选取目的基因片段在不同种菌株中存在差异,该技术方案无法适用于发酵乳杆菌Lactobacillus fermentum,也不能通过该技术方案得到发酵乳杆菌分离株的相应基因片段。此外,为了实现嗜热链球菌溯源,该技术方案采用分散选取的10个单拷贝等位基因进行比对,序列比对量庞大,因为实现效率较低。
技术实现要素:
本发明的目的是克服现有技术不足,提供一种发酵乳杆菌的快速溯源方法,以及实现该方法的看家基因联合序列及该序列的构建方法。
本发明的思路是从发酵乳杆菌的大量看家基因中选取部分特定基因为靶点,通过设计引物、PCR扩增获得含有dnaA、pyrG、rpoB、recA、dnaK和murC看家基因片段的联合序列,并通过测序、构建系统发育树实现发酵乳杆菌的快速溯源分析。
为此,本发明提供含有发酵乳杆菌基因组的联合基因序列在发酵乳杆菌溯源中的应用,所述联合基因序列包括dnaA、pyrG、rpoB、recA、dnaK和murC共6个看家基因。
上述联合基因序列的构建方法包括以下步骤:
(1)根据发酵乳杆菌的看家基因dnaA、pyrG、rpoB、recA、dnaK和murC设计特异性扩增引物;
(2)利用步骤(1)的引物分别通过PCR扩增发酵乳杆菌模板DNA,分别得到各看家基因片段,
扩增体系:
PCR mix(10μl,含有5U/μL Taq酶0.08μl),10×PCR buffer(Mg2+free)1μl,2.5mmol/L dNTPs 0.8μL,25mmo1/L Mg2+0.8μL,上下游引物各0.4μL,DNA模板(10-50ng/μl)1μl,dd H2O 5.52μL;
扩增条件:
95℃变性5min;循环30次:95℃变性1min,50℃退火45s,72℃延伸1min,然后72℃延伸10min,4℃保存;
(3)核苷酸序列测定
扩增产物经琼脂糖凝胶电泳检测合格,按照ABI 3730XL测序平台技术流程进行双向测序;
(2.4)拼接序列
将测序所得的双向序列导入软件DNAstar的Seqman模块中,结合序列峰图进行序列比对拼接及单碱基校对;然后采用6.0软件包分析,经Clustal W比对,并按照dnaA-pyrG-rpoB-recA-dnaK-murC顺序拼接得到所述联合基因序列。
其中,6个看家基因位置信息和引物信息如表1:
表1
注:a位置信息为发酵乳杆菌Lactobacillus fermentum F-6基因组的位置。b位置信息为看家基因中的碱基位置。
根据现有技术的记载,发酵乳杆菌的看家基因约有600-1000个,它们关系到菌株基础代谢和合成以及生长,但目前尚未有报道揭示哪些基因能用于揭示菌株来源。本发明在已知信息的基础上,选择同时具有保守性和突变位点的基因,通过设计引物扩增得到较短的基因片段,构造联合基因序列以验证快速筛选方法。
本发明的发酵乳杆菌快速溯源方法包括以下步骤:
(1)取初步鉴定为发酵乳杆菌的菌株,经镜检后对确定为纯培养物的菌株提取其基因组DNA;
(2)根据前述的构建方法,分别构建构建步骤(1)各菌株的联合基因序列;
(3)采用MEGA6.0软件包分析,经Clustal W比对、对步骤(2)的各联合基因序列构建邻接法系统发育树,通过发育树获得发酵乳杆菌菌株的溯源分析;
(4)将在系统发育树中处于同一分支的菌株判断为同一来源。
其中,步骤(1)包括:
(1.1)取保存于安瓿管中的冻干菌粉,置于脱脂乳中活化复壮,对菌液涂片镜检,将镜检结果为革兰氏阳性的纯培养物继续传代培养至第三代;
(1.2)取上述三代纯培养物菌液1mL进行洗涤处理,将洗脱的菌体置于1.5mL离心管中,并加入1mL PBS缓冲液,立即置于液氮中完全冻结,取出后放入65℃水浴中融化,反复冻融3次,加0.1mL 10wt%SDS和10.0μL 10mg/mL蛋白酶K,于37℃恒温摇床中200r/min摇动2h,室温下12000g离心10min,收集上清液转移至另一离心管中;上清液与等体积的氯仿于12000g离心10min,吸取上清液转移至另一离心管中进行酚氯仿抽提2次,随后加入0.1倍体积的醋酸钠和1倍体积的冰异丙醇沉淀总DNA,最后用70vol%乙醇洗涤沉淀2次,回溶备用;
(1.3)使用ND-1000型微量紫外分光光度计检测所提取DNA的浓度和纯度,用0.8%的琼脂糖凝胶对发酵乳杆菌基因组DNA进行电泳检测,电泳图应条带清晰,无拖尾。
本发明的快速筛选方法能够有效实现发酵乳杆菌多个分离株的溯源,通过发育树的分支情况,处于同一发育树的菌株可以被鉴定为同一来源。
【附图说明】
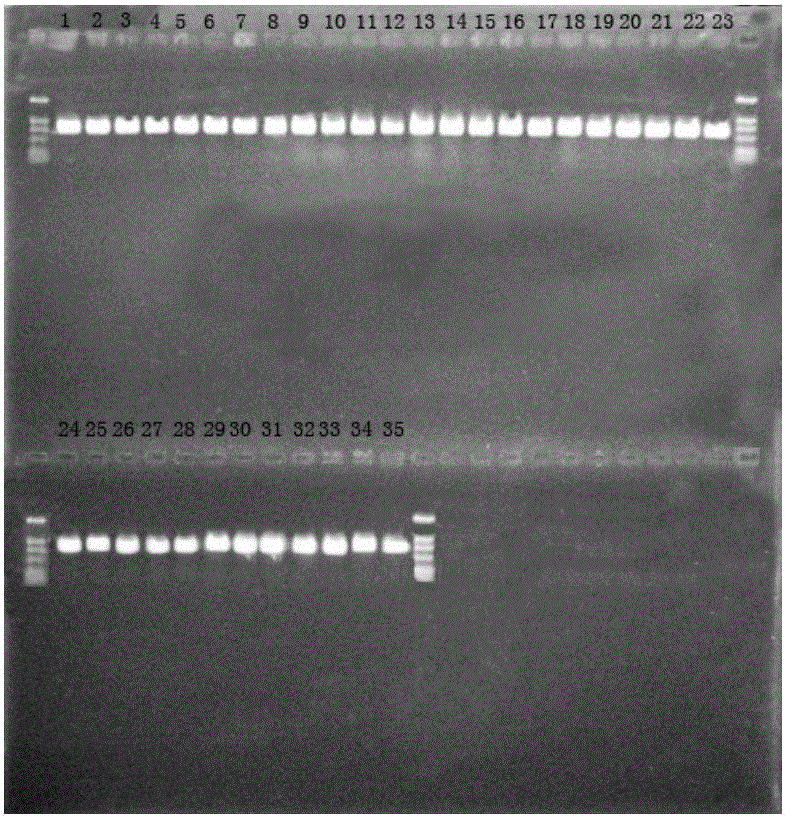
图1为表2的分离株的dnaA基因PCR扩增产物的琼脂糖凝胶电泳图;
图2为表2的分离株的pyrG基因PCR扩增产物的琼脂糖凝胶电泳图;
图3为表2的分离株的rpoB基因PCR扩增产物的琼脂糖凝胶电泳图;
图4为表2的分离株的recA基因PCR扩增产物的琼脂糖凝胶电泳图;
图5为表2的分离株的dnaK基因PCR扩增产物的琼脂糖凝胶电泳图;
图6为表2的分离株的murC基因PCR扩增产物的琼脂糖凝胶电泳图;
图7为实施例3的发育树图。
【具体实施方式】
以下实施例用于非限制性地解释本发明的技术方案。
在本发明中,如无特殊说明,用于解释浓度的“%”均为重量百分比、“份”均为重量份。
在本发明中,涉及以下培养基:
脱脂乳培养基:取10g脱脂乳粉,复溶于100mL蒸馏水,115℃灭菌15min,备用。
实施例1菌株的活化及DNA提取
选取从内蒙古西部地区、内蒙古呼和浩特市托克托县双河镇、云南省洱源县右所镇、蒙古国布尔干省阿日希亚图苏木、蒙古国肯特省巴音敖包苏木等地采集的酸粥、酸马奶、乳扇酸乳清和酸牛奶等样品中分离到的35株发酵乳杆菌,菌株分离信息见表2。
表2 Lactobacillus fermentum分离株来源信息
分别对菌株进行活化:将保存于安瓿管中的冻干菌粉,置于脱脂乳中活化复壮(37℃,24h),对菌液涂片镜检,将镜检结果为革兰氏阳性的纯培养物继续传代培养至第三代。
采用液氮冻融-CTAB法分别提取菌株DNA:取1.0mL第三代菌液洗涤处理,将洗脱的菌体于1.5mL离心管中,并加入1mL的PBS缓冲液,立即置于液氮中完全冻结,取出后放入65℃水浴中融化(约5min),反复冻融3次,加0.1mL 10%SDS和10.0μL 10mg/mL蛋白酶K,于37℃恒温摇床中200r/min摇动2h,室温下12000g离心10min,收集上清液转移至另一离心管中。上清液与等体积的氯仿于12000g离心10min(为了使沉淀、水相和有机相分层),吸取上清液转移至另一离心管中进行酚氯仿抽提2次,随后加入0.1倍体积的醋酸钠和1倍体积的冰异丙醇沉淀总DNA,最后用70%乙醇洗涤沉淀2次,回溶备用。
实施例2联合序列的构建
2.1设计引物
以发酵乳杆菌Lactobacillus fermentum F-6基因组(genbank编号CP005958)为模板,从大量已知的发酵乳杆菌的看家基因出发,通过比较基因组学分析结果和看家基因选择依据,考虑看家基因必须是与核心代谢密切相关的,且选取的基因要尽量覆盖整个基因组才能更准确地反映菌株间的进化关系,两个相邻的管家基因之间的距离也不应太近以避免遗传连锁性的影响,发明人经过多次试验,最终选取与发酵乳杆菌核心代谢相关的dnaA、pyrG、rpoB、recA、dnaK和murC等6个看家基因为靶点。采用Primer Premier 5.0软件设计特异性扩增引物,人工合成各引物。
表3六个看家基因PCR扩增引物
2.2
利用上述引物通过PCR扩增实施例1的分离株的模板DNA,每株分离株分别得到六种看家基因片段。
扩增体系:PCR mix(10μl)含有(5U/μL Taq酶0.08μl),10×PCR buffer(Mg2+free)1μl,2.5mmol/L dNTPs 0.8μL,25mmo1/L Mg2+0.8μL,上下游引物各0.4μL,DNA模板(10–50ng/μl)1μl,dd H2O 5.52μL;
扩增条件:
94℃变性5min;循环30次:95℃变性1min,50℃退火45s,72℃延伸1min,然后72℃延伸10min,4℃保存;
2.3核苷酸序列测定
各分离株的扩增产物如图1-6所示,扩增产物经琼脂糖凝胶电泳检测合格,各泳道产物条带清晰明亮、无拖尾、无明显非特异性扩增,说明PCR扩增成功,其产物可用于后续实验。
按照ABI 3730XL测序平台技术流程进行双向测序,测序峰图文件无双峰或杂峰,表明可用于后续分析。
2.4数据分析
测序所得原始数据经DNASTAR软件SeqMan模块质控、拼接,获得单一序列,再将每株分离株的6个基因序列按dnaA-pyrG-rpoB-dnaK-recA-murC的顺序串联起来,得到35株分离株的联合基因序列。其中,作为示例,样品编号1和22的核酸序列如SEQ No.13-14所示,其余联合基因序列也通过相同的软件操作得到。
实施例3构建发育树
采用MEGA6.0软件包分析联合序列,经Clustal W比对,对实施例1分离得到的35株菌的联合序列进行两两比对,计算序列间的距离,构建距离矩阵,以距离矩阵反映各序列之间亲缘关系。
然后通过构建邻接法(Neighour-joining,N-J)构建系统发育树(见图7)。
图7的基于danA-pyrG-rpoB-recA-dnaK-murC的联合基因序列构建的系统发育树可以看出35株发酵乳杆菌株构建的系统发育树很清晰地分为2个分支,分别由分离自中国和蒙古国的发酵乳杆菌分离株组成,所得结果与采集来源完全一致,表明处于同一分支的菌株具有相同的采集来源。
进一步地,在中国分支中,分离自中国内蒙古西部地区酸粥样品的分离株IMAU70150、IMAU70151、IMAU70149、IMAU70148、IMAU70147、IMAU70146、IMAU70145、IMAU70075、IMAU70076、IMAU70152、IMAU70153和IMAU70154形成一个独有的聚类,识别为酸粥分离源。
分离自中国内蒙古东部自然发酵酸马奶样品的IMAU10692、IMAU10705和IMAU10687形成一个聚类结构,识别为酸马奶分离源。
分离自中国云南乳扇酸乳清样品中的IMAU50088、IMAU50008、IMAU50071、IMAU50074、IMAU50077和IMAU50078聚为一个类群,识别为乳扇分离源。
分离自蒙古国自然发酵酸牛奶样品的IMAU20700、IMAU20704、IMAU20698、IMAU20694、IMAU20439、IMAU20440、IMAU20442、IMAU20443、IMAU20651、IMAU20652、IMAU20263和IMAU20268形成一个大的聚类,识别为蒙古国酸牛奶分离源。
IMAU20045、IMAU20046分离自蒙古国前杭盖省塔日雅图苏木的酸马奶。IMAU20700、IMAU20704、IMAU20698、IMAU20694分离自蒙古国布尔干省阿日希亚图苏木的酸牛奶,IMAU20439、IMAU20440、IMAU20442、IMAU20443分离自蒙古国库苏古尔省库苏古尔湖的酸牛奶,IMAU20651、IMAU20652分离自蒙古国后杭盖省图布希日夫苏木酸牛奶,IMAU20263、IMAU20268分离自蒙古国肯特省巴音敖包苏木酸牛奶;IMAU50088菌株分离自中国云南省洱源县右所镇的乳扇酸乳清菌和IMAU50008、IMAU50071、IMAU50074、IMAU50077、IMAU50078菌株分离自中国云南省大理市上关镇。
以上聚类关系与表1的能够准确对应。可见,本发明的基于dnaA-pyrG-rpoB-dnaK-recA-murC的联合基因序列及应用该序列的筛选方法能快速分离不同来源的菌株,实现发酵乳杆菌的采集来源溯源,根据发育树关系明确不同来源地的发酵乳杆菌,为日后进一步的研究提供基础。
作为对照,同样以发酵乳杆菌Lactobacillus fermentum F-6基因组(genbank编号CP005958)为模板,考察以其他看家基因如pepX、clpX为靶点。采用Primer Premier 5.0软件设计特异性扩增引物。
以与实施例2相同的扩增体系及条件获得基因片段。类似地,经琼脂糖凝胶电泳检测合格,再按照ABI 3730XL测序平台技术流程进行双向测序,然后尝试不同的串联方式,得到对照的各分离株的联合基因序列。
与实施例3相同进行,采用MEGA6.0软件包分析联合序列,经Clustal W比对,对35株菌的序列进行两两比对,计算序列间的距离,构建距离矩阵,以距离矩阵反映各序列之间亲缘关系。
然后通过构建邻接法构建系统发育树。然而,由于基因选择不同,尽管均为看家基因,但同一采集来源的分离株在发育树中最终表现为无序分散在各分支中,无法与表1实现一一对应,因此不能实现有效溯源。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种植物乳杆菌及其在发酵腌腊肉制品中的应用
- 用布氏乳杆菌、黑曲霉和植物乳杆菌组合固态发酵木薯渣的方法
- 一株植物乳杆菌及其在发酵饲料中的应用
- 一株植物乳杆菌及其在生产发酵鹅肉中的应用
- 一种植物乳杆菌发酵虾头、虾壳回收蛋白质和甲壳素的方法
- 一种可预防胃溃疡的发酵乳杆菌Lactobacillus fermentum strain Lee及其用图
- 一种可预防胃溃疡的发酵乳杆菌Lactobacillus fermentum Lee及其保健用图
- 一种可预防胃溃疡的发酵乳杆菌Lactobacillus fermentum strain suo及其用图
- 高密度固态发酵培养乳杆菌的培养基及方法
- 一种植物乳杆菌处理甜菜汁制备发酵型甜菜浸膏的方法
- 用布氏乳杆菌、黑曲霉和植物乳杆菌组合固态发酵木薯渣的方法
- 一株植物乳杆菌及其在发酵饲料中的应用
- 一株植物乳杆菌及其在生产发酵鹅肉中的应用
- 一种植物乳杆菌发酵虾头、虾壳回收蛋白质和甲壳素的方法
- 一种可预防胃溃疡的发酵乳杆菌Lactobacillus fermentum strain Lee及其用图
- 一种可预防胃溃疡的发酵乳杆菌Lactobacillus fermentum Lee及其保健用图
- 一种可预防胃溃疡的发酵乳杆菌Lactobacillus fermentum strain suo及其用图
- 高密度固态发酵培养乳杆菌的培养基及方法
- 一种植物乳杆菌处理甜菜汁制备发酵型甜菜浸膏的方法
- 一种可预防胃溃疡的发酵乳杆菌Lactobacillus fermentum suo及其保健用图