噻吩类化合物、其制备方法与应用、钙钛矿太阳电池与流程
本发明属于钙钛矿太阳电池技术领域,尤其涉及一种噻吩类化合物、其制备方法与应用、钙钛矿太阳电池。
背景技术:
2009年,有机-无机杂化钙钛矿自作为光吸收材料首次被应用在太阳电池。2012年后,有关此类新型太阳电池的报道呈井喷式增长,至2016年实现了22.1%的能量转换效率,超过1cm2的器件实现了19.6%的能量转换效率(Science,2016,353,58-62)。与此同时,溶液加工的钙钛矿太阳电池,可通过roll-to-roll实现高效、大规模批量生产,相比于传统的硅基太阳电池,制造成本低、加工工艺简单,具有良好商业前景。
高效的钙钛矿太阳电池中最广泛使用的有机空穴输运材料主要为2,2′,7,7′-四[N,N-二(4-甲氧基苯基)氨基]-9,9′-螺二芴(Spiro-OMeTAD)。但是Spiro-OMeTAD化学结构复杂、合成路线长、价格昂贵,同时该材料空穴迁移率较低,通常需要采用双(三氟甲基磺酰亚胺)锂(LiTFSI)、叔丁基吡啶(TBP)以及双(三氟甲基磺酰亚胺)钴(FK209)进行p型掺杂来提高空穴迁移率,但这类掺杂会导致电池器件性能不稳定,同时材料费用昂贵。因此,发展新型低成本、高迁移率的空穴输运材料对于提高钙钛矿太阳电池光电性能以及加快商业化步伐都具有重要的意义。
技术实现要素:
有鉴于此,本发明所要解决的技术问题在于提供一种高迁移率的噻吩类化合物、其制备方法与应用。
本发明提供了一种噻吩类化合物,如式(I)所示:
其中-A-选自-S-、
R1与R1′各自独立地选自H、C1~C12烃基或C1~C12烷氧基;
R2、R3与R4各自独立地选自H、C1~C12烃基、C1~C12烷氧基或C1~C12烷氧基取代的苯基。
优选的,所述R1与R1′各自独立地选自H、C1~C6烃基或C1~C6烷氧基。
优选的,所述R1与R1′各自独立地选自H、C1~C3烃基或C1~C3烷氧基。
优选的,所述R2、R3与R4各自独立地选自H、C1~C6烃基、C1~C6烷氧基或C1~C6烷氧基取代的苯基。
优选的,所述R2、R3与R4各自独立地选自H、C1~C3烃基、C1~C3烷氧基或C1~C3烷氧基取代的苯基。
优选的,如式(I-1)~式(I-5)所示:
本发明还提供了一种噻吩类化合物的制备方法,包括:
将式(II)所示的化合物与式(III)所示的化合物进行反应,得到式(I)所示的噻吩类化合物;
其中-A-为-S-、
X选自卤原子;
R1与R1′各自独立地选自H、C1~C6烃基或C1~C6烷氧基;
R2、R3与R4各自独立地选自H、C1~C6烃基、C1~C6烷氧基或C1~C6烷氧基取代的苯基。
本发明还提供了一种噻吩类化合物作为空穴传输材料的应用。
本发明还提供了一种钙钛矿太阳电池,包括噻吩类化合物。
优选的,包括空穴传输层;所述空穴传输层噻吩类化合物。
本发明提供了一种噻吩类化合物、其制备方法与应用、钙钛矿太阳电池,该噻吩类化合物如式(I)所示,R1与R1′各自独立地为H、C1~C12烃基或C1~C12烷氧基;R2、R3与R4各自独立地为H、C1~C12烃基、C1~C12烷氧基或C1~C12烷氧基取代的苯基。与现有技术相比,本发明提供的噻吩类化合物为并三噻吩、噻吩并吡咯或双噻吩并环戊二烯与两个三芳胺共轭偶联的结构,使其空穴迁移率较高,进而提高使具有该噻吩类化合物制备而成的空穴输运层的钙钛矿太阳电池具有较高的能量转换效率。
实验结果表明,采用本发明制备的噻吩类化合物作为空穴传输层制备的钙钛矿太阳电池的能量转换效率可高达14.5%~16.6%。
附图说明
图1为本发明实施例5中得到的钙钛矿太阳电池的电压-电流曲线图;
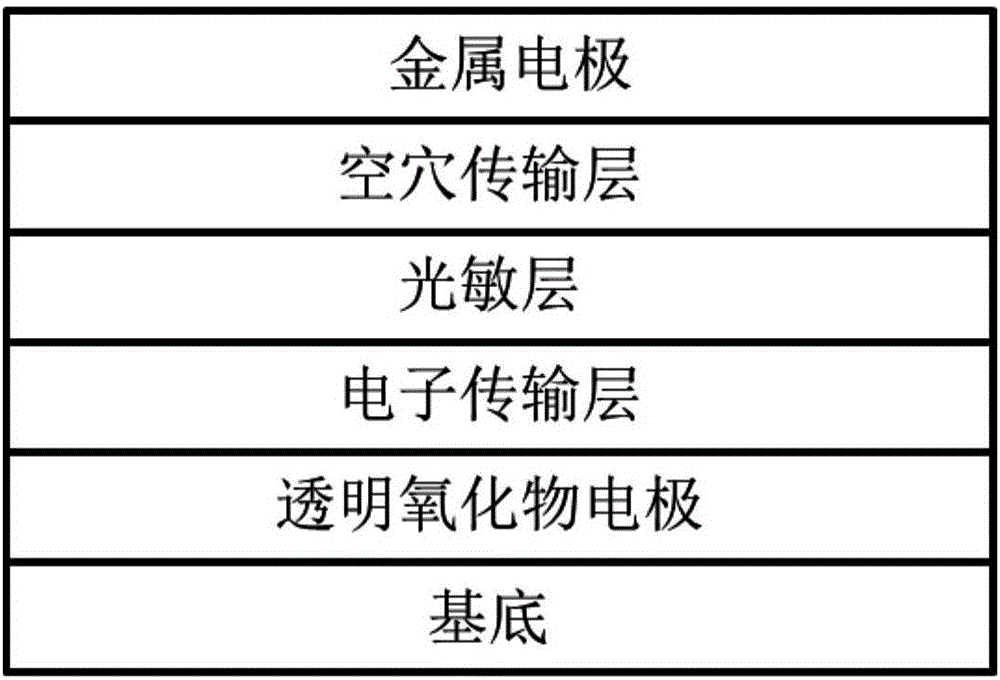
图2为本发明提供的钙钛矿太阳电池的器件结构示意图。
具体实施方式
本发明提供了一种噻吩类化合物,如式(I)所示:
其中-A-为-S-、
R1与R1′各自独立地为H、C1~C12烃基或C1~C12烷氧基,优选各自独立地为H、C1~C10烃基或C1~C10烷氧基,更优选各自独立地为H、C1~C6烃基或C1~C6烷氧基,再优选各自独立地为H、C1~C3烃基或C1~C3烷氧基;在本发明中R1与R1′可以同时为相同的取代基,也可为不同的取代基,并无特殊的限制,优选为同时为相同的取代基。
R2、R3与R4各自独立地为H、C1~C12烃基、C1~C12烷氧基或C1~C12烷氧基取代的苯基,优选各自独立地为H、C1~C8烃基、C1~C8烷氧基或C1~C8烷氧基取代的苯基,更优选各自独立地为H、C1~C6烃基、C1~C6烷氧基或C1~C6烷氧基取代的苯基,再优选各自独立地为H、C1~C3烃基、C1~C3烷氧基或C1~C3烷氧基取代的苯基。
在本发明中,所述烃基可为直链烃基也可为支链烃基,并无特殊的限制;所述烷氧基可为直链的烷氧基也可为支链烷氧基,并无特殊的限制。
在本发明中,所述噻吩类化合物最优选如式(I-1)~式(I-5)所示:
本发明提供的噻吩类化合物为并三噻吩、噻吩并吡咯或双噻吩并环戊二烯与两个三芳胺共轭偶联的结构,使其空穴迁移率较高,进而提高使具有该噻吩类化合物制备而成的空穴输运层的钙钛矿太阳电池具有较高的能量转换效率。
本发明还提供了一种上述噻吩类化合物的制备方法,包括:
将式(II)所示的化合物与式(III)所示的化合物进行反应,得到式(I)所示的噻吩类化合物;
其中-A-为-S-、
X为卤原子,优选为Cl或Br,更优选为Br;
R1为H、C1~C6烃基或C1~C6烷氧基;
R2、R3与R4各自独立地为H、C1~C6烃基、C1~C6烷氧基或C1~C6烷氧基取代的苯基。
所述R1、R2、R3与R4均同上所述,在此不再赘述。
按照本发明,所述反应的条件为本领域技术人员熟知的Suzuki偶联反应条件即可,并无特殊的限制。
本发明提供的基于稠环噻吩共轭单元的有机空穴传输材料,其结构更加简单,大大缩减了加工制备的路线。
本发明还提供了一种上述噻吩类化合物作为空穴传输材料的应用。
本发明还提供了一种钙钛矿太阳电池,包括上述的噻吩类化合物。
按照本发明,所述钙钛矿太阳电池优选包括空穴传输层,所述空穴传输层包括上述噻吩类化合物;所述空穴传输层的厚度优选为10~150nm。
在本发明中,所述钙钛矿太阳电池的器件结构参见图2,依次包括基底、透明氧化物电极、电子传输层、光敏层、空穴传输层与金属电极。
其中,所述基底为本领域技术人员熟知的基底即可,并无特殊的限制,本发明中优选为玻璃、石英、柔性PET或PEN。
所述的透明氧化物电极为本领域技术人员熟知的透明氧化物电极即可,并无特殊限制,本发明中优选为氟掺氧化锡(FTO)或氧化铟锡(ITO)。
所述电子传输层为本领域技术人员熟知的电子传输层即可,并无特殊的限制,本发明中优选为TiO2、PC61BM、PC71BM或ZnO。
所述光敏层为本领域技术人员熟知光敏层即可,并无特殊的限制,本发明中优选为具有以下化学结构式Csx(FA0.83MA0.17)(1-x)Pb(Br0.17I0.83)3,其中0≤x≤0.1。
所述金属电极为本领域技术人员熟知的金属电极即可,并无特殊的限制,本发明中优选为金、银、镁、铝或钙;所述金属电极的厚度优选为100~400nm。
所述钙钛矿电池的制备方法为本领域技术人员熟知的制备方法即可,并无特殊的限制,以电子传输层为TiO2为例本发明优选按照以下方法进行:
将表面刻蚀有图案的基底清洗,烘干,再用紫外臭氧机处理;将处理后的基底置于40mM的TiCl4-盐酸水溶液中,在70℃下水解1.5h,并用去离子水、乙醇冲洗;接着,在基底上旋涂TiO2的乙醇溶液,于热板上120℃退火10min后,在空气流中450℃煅烧30min,其中TiO2浆料和乙醇的质量比为1:3.5,旋涂条件4000r,20s;然后在TiO2衬底上旋涂LiTFSI的乙腈溶液,浓度0.1M,旋涂条件3000r,30s,之后在空气流中450℃煅烧30min;然后,一步法旋涂钙钛矿前驱溶液制备钙钛矿薄膜,100℃退火30min,溶液配方:1M FAI,1.1M PbI2,0.2MMABr,0.2MPbBr2,0.063MCsI溶解于DMSO和DMF体积比为1:4的混合溶剂中,旋涂条件:1000r,10s,6000r,30s,其中在第二阶段旋涂最后5s的时候用150uL的氯苯洗涤旋转中的衬底。最后,在钙钛矿表面旋涂空穴输运材料,溶液配方:60mM本发明上述的噻吩类化合物溶解于CB溶液中,旋涂条件:6000r,30s。最后,真空蒸镀上金属电极电极,得到钙钛矿太阳电池。
下面将结合本发明实施例的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
以下实施例中,所用化学试剂均从安耐吉化学试剂公司购得;化合物1,2,6-二溴并三噻吩根据文献(R.Kondo,T.Yasuda,Y.S.Yang,J.Y.Kim,C.Adachi,J.Mater.Chem.,2012,22,16810.)合成得到,化合物2,4-(4-甲氧基苯基)-4H-联噻吩[3,2-b:2',3'-d]吡咯根据文献(H.L.Wong,C.C.Ko,W.H.Lam,N.Zhu,V.W.W.Yam,Chem.Eur.J.2009,15,10005.)合成得到,化合物4,2,6-二溴-4,4-二甲基-4H-环戊烯[1,2-b:5,4-b']联噻吩根据文献(H.Hanamura,R.Haneishi,N.Nemoto,Tetrahedron Letters,2011,52,4039.)合成得到。
实施例1
在干燥的Schlenk反应瓶中,分别加入1g中间体1、4.87g 4-甲氧基-N-(4-甲氧基苯基)-N-(4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)苯基)胺、3.59g碳酸钠,然后加入50mL甲苯、50mL水溶解,氩气保护下加入130mg Pd(PPh3)4,升温至回流,磁力搅拌下反应过夜。
TLC检测反应完毕后,待反应体系冷却至室温,加入水,并用乙酸乙酯萃取三次,合并有机相,有机相利用无水硫酸钠干燥,滤去干燥剂,旋干后用甲苯/石油醚(体积比4∶1)作为展开剂柱层析,得到黄色固体粉末目标产物即式(I-1)所示的噻吩类化合物1.5g。
采用核磁共振、质谱分析和元素分析法对得到的目标产物式I-1进行结构表征,高分辨质谱分析结果:803.02154。元素分析结果:C,71.80;H,4.75,N,3.48。
核磁共振表征数据如下:
1H NMR(400MHz,THF-d8)δ:7.51(s,2H),7.46(d,J=8.6Hz,4H),7.06(d,J=8.8Hz,8H),6.90(d,J=8.7Hz,4H),8.86(d,J=8.8Hz,8H),3.76(s,12H)。
13C NMR(100MHz,THF-d8)δ:157.69,149.89,146.17,142.76,141.55,130.03,127.83,127.51,127.04,121.18,116.13,115.71,55.78。实验结果表明,本发明制备得到了式(I-1)所示的噻吩类化合物。
实施例2
2.1 目标产物式(I-2)所示的噻吩类化合物的合成
在干燥的Schlenk反应瓶中,加入720mg中间体2溶解于10mL四氢呋喃中,冰浴完全后,加入1.12g氮溴代琥珀亚酰胺,磁力搅拌下反应30min,TLC检测反应完毕。加入20mL水,反应混合液用氯仿萃取,合并有机相,有机相用无水硫酸钠干燥,滤除干燥剂,旋干溶剂,所得白色固体粉末直接用于下步反应。
在干燥的Schlenk反应瓶中,加入上述反应所得产物600mg,再加入1.614g 4-甲基-N-(4-甲基苯基)-N-(4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)苯基)胺、1.43g磷酸钾,然后加入20mL二氧六环、4mL水溶解,氩气保护下加入7.26mg Pd(OAC)2、13.27mg Sphos升温至回流,磁力搅拌下反应过夜。
TLC检测反应完毕后,待反应体系冷却至室温,加入水,并用乙酸乙酯萃取三次,合并有机相,有机相利用无水硫酸钠干燥,滤去干燥剂,旋干后用甲苯/石油醚(体积比1:2)作为展开剂柱层析,得到黄色固体粉末目标产物即式(I-2)所示的噻吩类化合物700mg。
采用核磁共振、质谱分析和元素分析法对得到的目标产物式(I-2)所示的噻吩类化合物进行结构表征,高分辨质谱分析结果:827.29863。元素分析结果:C,79.78;H,5.48,N,5.06。
核磁共振表征数据如下:
1H NMR(400MHz,THF-d8)δ:7.59(d,J=8.7Hz,2H),7.49(d,J=8.4Hz,4H),7.32(s,2H),7.11(d,J=8.7Hz,2H),7.06(d,J=8.2Hz,8H),6.97(d,J=8.3Hz,12H),3.86(s,3H),2.29(s,12H)。
13C NMR(100MHz,THF-d8)δ:159.56,148.60,146.35,145.58,143.18,133.76,133.56,130.14,125.69,123.73,116.09,115.93,107.97,55.98,21.02。实验结果表明,本发明制备得到了具有式(I-2)所示的噻吩类化合物。
2.2 目标产物式(I-3)所示的噻吩类化合物的合成
在干燥的Schlenk反应瓶中,加入600mg中间体2溶解于10mL四氢呋喃中,冰浴完全后,加入933mg氮溴代琥珀亚酰胺,磁力搅拌下反应30min,TLC检测反应完毕。加入20mL水,反应混合液用氯仿萃取,合并有机相,有机相用无水硫酸钠干燥,滤除干燥剂,旋干溶剂,所得白色固体粉末直接用于下步反应。
在干燥的Schlenk反应瓶中,加入上述反应所得产物598mg,再加入1.994g 4-甲氧基-N-(4-甲氧基苯基)-N-(4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)苯基)胺、1.43g磷酸钾,然后加入20mL二氧六环、4mL水溶解,氩气保护下加入12mg Pd(OAC)2、22mg Sphos升温至回流,磁力搅拌下反应过夜。
TLC检测反应完毕后,待反应体系冷却至室温,加入水,并用乙酸乙酯萃取三次,合并有机相,有机相利用无水硫酸钠干燥,滤去干燥剂,旋干后用甲苯/石油醚(体积比1:1)作为展开剂柱层析,得到橙色固体粉末目标产物即式(I-3)所示的噻吩类化合物820mg。
采用核磁共振、质谱分析和元素分析法对得到的目标产物式(I-3)所示的噻吩类化合物进行结构表征,高分辨质谱分析结果:891.27805。元素分析结果:C,74.06;H,5.08,N,4.70。
核磁共振表征数据如下:
1H NMR(400MHz,THF-d8)δ:7.58(d,J=8.7Hz,2H),7.44(d,J=8.5Hz,4H),7.28(s,2H),7.11(d,J=8.8Hz,2H),7.03(d,J=8.8Hz,8H),6.88(d,J=8.6Hz,12H),3.86(s,3H),2.29(s,12H)。
13C NMR(100MHz,THF-d8)δ:159.52,157.48,149.27,145.49,143.34,141.80,133.84,128.94,126.75,125.65,121.66,115.92,115.64,107.58,55.98,55.76。实验结果表明,本发明制备得到了具有式(I-3)所示的噻吩类化合物。
实施例3
反应路线如下:
3.1 目标产物式(I-4)所示的噻吩类化合物的合成
在干燥的Schlenk反应瓶中,加入1.292g中间体2溶解于20mL四氢呋喃中,冰浴完全后,加入2.43g氮溴代琥珀亚酰胺,磁力搅拌下反应30min,TLC检测反应完毕。加入20mL水,反应混合液用氯仿萃取,合并有机相,有机相用无水硫酸钠干燥,滤除干燥剂,旋干溶剂,所得白色固体粉末直接用于下步反应。
在干燥的Schlenk反应瓶中,加入上述反应所得产物1.3g,再加入3.14g4-甲基-N-(4-甲基苯基)-N-(4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)苯基)胺、3.79g磷酸钾,然后加入20mL二氧六环、4mL水溶解,氩气保护下加入32mg Pd(OAC)2、59mg Sphos升温至回流,磁力搅拌下反应过夜。
TLC检测反应完毕后,待反应体系冷却至室温,加入水,并用乙酸乙酯萃取三次,合并有机相,有机相利用无水硫酸钠干燥,滤去干燥剂,旋干后用甲苯/石油醚(体积比1:2)作为展开剂柱层析,得到橙色固体粉末目标产物即式(I-4)所示的噻吩类化合物1.5g。
采用核磁共振、质谱分析和元素分析法对得到的目标产物式(I-4)所示的噻吩类化合物进行结构表征,高分辨质谱分析结果:748.29230。元素分析结果:C,81.77;H,5.93,N,3.75。
核磁共振表征数据如下:
1H NMR(400MHz,THF-d8)δ:7.46(d,J=8.4Hz,4H),7.29(s,2H),7.07(d,J=8.1Hz,8H),6.97(d,J=8.3Hz,12H),2.29(s,12H),1.50(s,6H)。
13C NMR(100MHz,THF-d8)δ:162.16,148.48,146.35,145.90,134.78,133.56,129.88,125.65,123.72,117.20,110.67,46.68,21.04。实验结果表明,本发明制备得到了式(I-4)所示的噻吩类化合物。
3.2 目标产物式(I-5)所示的噻吩类化合物的合成
在干燥的Schlenk反应瓶中,加入1.292g中间体2溶解于20mL四氢呋喃中,冰浴完全后,加入2.43g氮溴代琥珀亚酰胺,磁力搅拌下反应30min,TLC检测反应完毕。加入20mL水,反应混合液用氯仿萃取,合并有机相,有机相用无水硫酸钠干燥,滤除干燥剂,旋干溶剂,所得白色固体粉末直接用于下步反应。
在干燥的Schlenk反应瓶中,加入上述反应所得产物700mg,再加入2.86g4-甲氧基-N-(4-甲氧基苯基)-N-(4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)苯基)胺、2.04g磷酸钾,然后加入20mL二氧六环、4mL水溶解,氩气保护下加入17mg Pd(OAC)2、32mg Sphos升温至回流,磁力搅拌下反应过夜。
TLC检测反应完毕后,待反应体系冷却至室温,加入水,并用乙酸乙酯萃取三次,合并有机相,有机相利用无水硫酸钠干燥,滤去干燥剂,旋干后用甲苯/石油醚(体积比4:1)作为展开剂柱层析,得到橙色固体粉末目标产物即式(I-5)所示的噻吩类化合物820mg。
采用核磁共振、质谱分析和元素分析法对得到的目标产物式(I-5)所示的噻吩类化合物进行结构表征,高分辨质谱分析结果:812.27266。元素分析结果:C,75.34;H,5.46,N,3.44。
核磁共振表征数据如下:
1H NMR(400MHz,THF-d8)δ:7.41(d,J=8.1Hz,4H),7.24(s,2H),7.03(d,J=8.7Hz,8H),6.89-6.83(m,12H),3.76(s,12H),1.49(s,6H)。
13C NMR(100MHz,THF-d8)δ:162.05,157.49,149.15,146.03,141.79,134.49,128.67,127.55,126.63,121.62,116.79,115.65,110.73,55.76,46.45。实验结果表明,本发明制备得到了式(I-5)所示的噻吩类化合物。
实施例4
将表面刻蚀有图案的FTO透明导电玻璃依次用清洗剂、去离子水、丙酮、乙醇超声清洗10min后,烘干,再用紫外臭氧机处理15min。
首先,将FTO玻璃置于40mM的TiCl4-盐酸水溶液中,在70℃下水解1.5h,并用去离子水、乙醇冲洗;接着,在基底上旋涂TiO2的乙醇溶液,于热板上120℃退火10min后,在空气流中450℃煅烧30min,其中TiO2浆料和乙醇的质量比为1:3.5,旋涂条件4000r,20s;然后在TiO2衬底上旋涂LiTFSI的乙腈溶液,浓度0.1M,旋涂条件3000r,30s,之后在空气流中450℃煅烧30min。其次,一步法旋涂钙钛矿前驱溶液制备钙钛矿薄膜,100℃退火30min,溶液配方:1M FAI,1.1M PbI2,0.2M MABr,0.2MPbBr2,0.063MCsI溶解于DMSO和DMF体积比为1:4的混合溶剂中,旋涂条件:1000r,10s,6000r,30s,其中在第二阶段旋涂最后5s的时候用150uL的氯苯洗涤旋转中的衬底。最后,在钙钛矿表面旋涂空穴传输材料,溶液配方:60mM式(I-1)所示的噻吩类化合物的空穴输运材料溶解于氯苯溶液中,旋涂条件:6000r,30s。最后,真空蒸镀上60nm厚的Au电极,得到钙钛矿太阳电池。
在光照强度为100mW/cm2的AM1.5模拟太阳光下照射下,测试该器件的电压-电流曲线,从中得到开路电压为1.06V,短路电流密度为20.44mA/cm2,填充因子为0.715,能量转换效率为15.5%。
实施例5
将表面刻蚀有图案的FTO透明导电玻璃依次用清洗剂、去离子水、丙酮、乙醇超声清洗10min后,烘干,再用紫外臭氧机处理15min。
首先,将FTO玻璃置于40mM的TiCl4-盐酸水溶液中,在70℃下水解1.5h,并用去离子水、乙醇冲洗;接着,在基底上旋涂TiO2的乙醇溶液,于热板上120℃退火10min后,在空气流中450℃煅烧30min,其中TiO2浆料和乙醇的质量比为1:3.5,旋涂条件4000r,20s;然后在TiO2衬底上旋涂LiTFSI的乙腈溶液,浓度0.1M,旋涂条件3000r,30s,之后在空气流中450℃煅烧30min。其次,一步法旋涂钙钛矿前驱溶液制备钙钛矿薄膜,100℃退火30min,溶液配方:1MFAI,1.1MPbI2,0.2MMABr,0.2MPbBr2,0.063MCsI溶解于DMSO和DMF体积比为1:4的混合溶剂中,旋涂条件:1000r,10s,6000r,30s,其中在第二阶段旋涂最后5s的时候用150uL的氯苯洗涤旋转中的衬底。最后,在钙钛矿表面旋涂空穴传输材料,溶液配方:60mM式(I-3)所示的噻吩类化合物溶解于CB溶液中,旋涂条件:6000r,30s。最后,真空蒸镀上60nm厚的Au电极,得到钙钛矿太阳电池。
本发明在100mW/cm2模拟AM1.5G太阳光下,测试该器件的电压-电流曲线,如图1所示,从图中得到开路电压为1.06V,短路电流密度为20.56mA/cm2,填充因子为0.761,能量转换效率为16.6%。
实施例6
将表面刻蚀有图案的FTO透明导电玻璃依次用清洗剂、去离子水、丙酮、乙醇超声清洗10min后,烘干,再用紫外臭氧机处理15min。
首先,将FTO玻璃置于40mM的TiCl4-盐酸水溶液中,在70℃下水解1.5h,并用去离子水、乙醇冲洗;接着,在基底上旋涂TiO2的乙醇溶液,于热板上120℃退火10min后,在空气流中450℃煅烧30min,其中TiO2浆料和乙醇的质量比为1:3.5,旋涂条件4000r,20s;然后在TiO2衬底上旋涂LiTFSI的乙腈溶液,浓度0.1M,旋涂条件3000r,30s,之后在空气流中45℃煅烧30min。其次,一步法旋涂钙钛矿前驱溶液制备钙钛矿薄膜,100℃退火30min,溶液配方:1MFAI,1.1MPbI2,0.2MMABr,0.2M PbBr2,0.063MCsI溶解于DMSO和DMF体积比为1:4的混合溶剂中,旋涂条件:1000r,10s,6000r,30s,其中在第二阶段旋涂最后5s的时候用150uL的氯苯洗涤旋转中的衬底。最后,在钙钛矿表面旋涂空穴传输材料,溶液配方:60mM式(I-5)所示的噻吩类化合物溶解于氯苯溶液中,旋涂条件:6000r,30s。最后,真空蒸镀上60nm厚的Au电极,得到钙钛矿太阳电池。
在光照强度为100mW/cm2的AM1.5模拟太阳光下照射下,测试该器件的电压-电流曲线,从中得到开路电压为1.01V,短路电流密度为20.69mA/cm2,填充因子为0.694,能量转换效率为14.5%。
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!