产乳酸微生物及使用其生产乳酸的方法与流程
本申请涉及一种产乳酸酵母属微生物和一种使用其生产乳酸的方法。
背景技术:
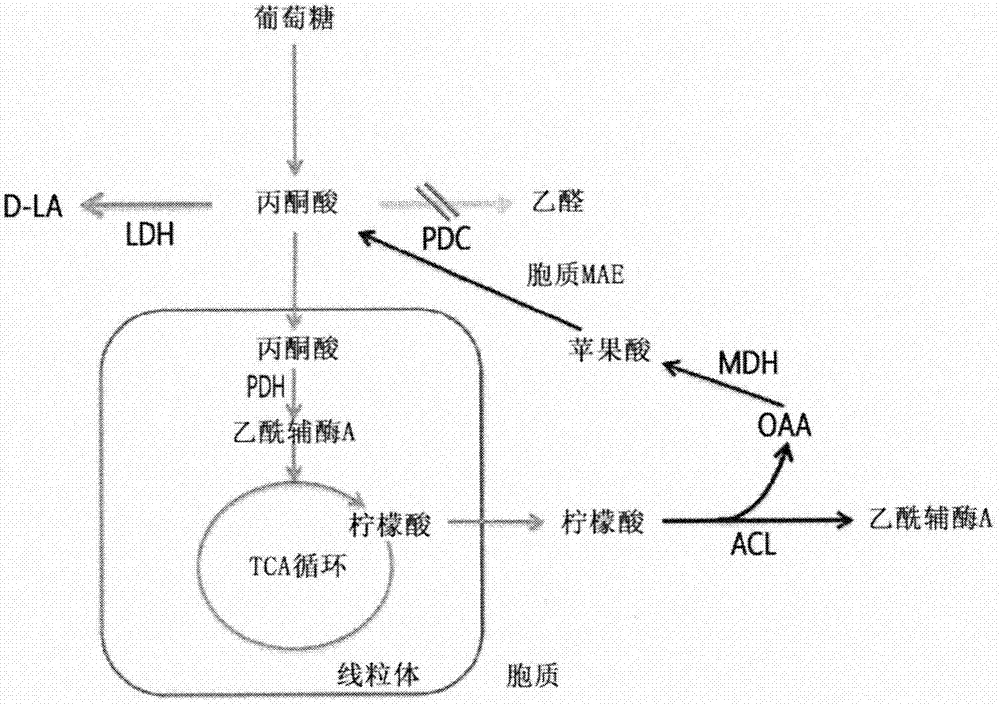
:乳酸在包括食品、医药、化妆品等在内的工业中具有广泛的应用。近来,由于使用乳酸作为聚乳酸的单体,因此需求显著增加。生产乳酸的方法包括传统化学合成和以碳水化合物作为底物的生物发酵过程,后者近来受到青睐。通常,在基于酵母的产乳酸微生物中,乳酸脱氢酶(LDH)和丙酮酸脱羧酶(PDC)竞争使用丙酮酸作为底物。在这种情况下,有必要使PDC的功能最小化以通过LDH生成更多乳酸(LA),来使丙酮酸的生产率最大化。在酿酒酵母(Saccharomycescerevisiae)中,PDC以三种不同形式的同工酶存在,即PDC1、PDC5和PDC6。因此,为使乳酸产量最大化,可以使用制备同时三重缺失PDC1、PDC5和PDC6的菌株的方法。然而,虽然PDC活性的失活可以增加乳酸的产率,但其也具有缺点,在于其降低生产能力并且菌株不能平稳地生长(欧洲专利号EP2041264),因此难以产生期望量的乳酸。另外,与原核生物如细菌不同,酵母如酵母属不能精确地预测是否可以通过各种细胞器和各种有机系统的简单基因操作来生产期望量的乳酸。技术实现要素:技术问题本发明人已努力开发了增加乳酸生产率和生产量,同时保持产乳酸微生物平稳生长的方法,结果是已证实增强乙酰辅酶A的供给路径和从草酰乙酸(OAA)到丙酮酸的供给路径提高了乳酸的生产量,从而完成了本申请。技术方案本申请的一个目的是提供一种产乳酸酵母属微生物。本申请的另一个目的是提供一种使用所述微生物生产乳酸的方法。有益效果本申请的经修饰的产乳酸菌株使得PDC活性失活、引入外源ACL活性并增强丙酮酸的生物合成途径,其与常规菌株相比,通过最小化产生醇的发酵和阻断乳酸分解途径而具有优异的乳酸发酵产率和生产力。因此,产乳酸菌株的生长增加,并且本申请的经修饰的产乳酸菌株可以被广泛地用于提高使用乳酸作为原材料制备的多种产品的生产力。由此生产的乳酸可作为多种产品的原材料提供。附图说明图1为经由存在于酵母微生物中的PEP羧激酶(PCK1,EC4.1.1.49)和丙酮酸激酶(PYK2,EC2.7.1.40)的过表达来增强丙酮酸的生物合成的途径,并示出了示意图,该示意图说明了本申请的通过引入外源ACL及增强PCK和PYK途径来提高乳酸生产力的策略。图2为通过增强存在于酵母微生物中的苹果酸脱氢酶2(MDH2,EC1.1.1.37)和胞质苹果酸酶1(MAE1,EC1.1.1.38)来增强丙酮酸的生物合成的途径,并示出了示意图,该示意图说明了本申请的通过引入外源ACL及经由MDH和胞质MAE途径来提高乳酸生产力的策略。具体实施方式为了实现上述目的,在一个方面,本申请提供了一种产乳酸酵母属微生物,其中所述微生物经修饰以使得丙酮酸脱羧酶(PDC)的活性与其内源活性相比失活,以引入ATP-柠檬酸裂解酶(ACL)的活性,并以与其内源生物合成途径相比增强丙酮酸生物合成途径。产乳酸酵母属微生物可以是这样的微生物,其中,与未经修饰的菌株相比,提高了乳酸发酵产率和/或增加了酵母属微生物体的生长(增加具有生产力的微生物的生长)和/或提高了乳酸生产力,在未经修饰的菌株中未引入ACL活性,和/或相对于内源生物合成途径,丙酮酸生物合成途径的未增强。如本文所使用的,术语“乳酸(LA)”指由C2H4OHCOOH表示的有机酸。当这样的乳酸通过化学合成生产时,乳酸以其中D-型乳酸和L-型乳酸以50/50比率混合的外消旋混合物形式产生,并且不能控制组成比。因此,当制备聚乳酸时,乳酸变为具有低熔点的非晶聚合物,并因此在开发其用途中有诸多限制。相比之下,当通过生物发酵方法使用微生物生产乳酸时,根据所使用的细菌或被引入其中的乳酸脱氢酶(LDH),可以选择性地生产D-型乳酸和L-型乳酸。如本文所使用的,术语“产乳酸微生物”指在本申请中具有乳酸生产力、可以将糖转化为乳酸的微生物菌株,例如可以包括任何酵母微生物而没有任何限制,只要其包括本申请的乳酸合成途径和乙酰辅酶A合成途径即可。根据其形状,酵母微生物可以被分类为酵母属、毕赤酵母(Pichia)属、假丝酵母(Candida)属和复膜孢酵母(Saccharomycopsis)属,具体而言,本申请中可以使用包括多种种类的酵母属微生物,只要该微生物可以生产乳酸即可。具体而言,酵母属微生物可为选自贝酵母(Saccharomycesbayanus)、布拉酵母(Saccharomycesboulardii)、Saccharomycesbulderi、Saccharomycescariocanus、Saccharomycescariocus、酿酒酵母、薛瓦酵母(Saccharomyceschevalieri)、Saccharomycesdairenensis、葡萄酒酵母(Saccharomycesellipsoideus)、Saccharomyceseubayanus、少孢酵母(Saccharomycesexiguus)、佛罗伦糖化酵母(Saccharomycesflorentinus)、克鲁维酵母(Saccharomyceskluyveri)、Saccharomycesmartiniae、Saccharomycesmonacensis、诺地酵母(Saccharomycesnorbensis)、奇异酵母(Saccharomycesparadoxus)、巴氏酵母(Saccharomycespastorianus)、Saccharomycesspencerorum、图列茨酵母(Saccharomycesturicensis)、单孢酵母(Saccharomycesunisporus)、葡萄汁酵母(Saccharomycesuvarum)和Saccharomyceszonatus,更具体而言,选自酿酒酵母。本申请的产乳酸酵母属微生物可以是这样的微生物,其中所述微生物经修饰以使得丙酮酸脱羧酶(PDC)的活性与其内源活性相比失活,以引入ATP-柠檬酸裂解酶的活性,并与其内源生物合成途径相比增强丙酮酸生物合成途径。具体而言,产乳酸酵母属微生物可以是这样的微生物,其中所述微生物经修饰(i)以使得丙酮酸脱羧酶(PDC)的活性与其内源活性相比失活,(ii)以引入ATP-柠檬酸裂解酶的活性,和(iii)以与其内源生物合成途径相比增强丙酮酸生物合成途径,并经进一步修饰(iv)以引入乳酸脱氢酶(LDH)的活性,(v)以使得乙醇脱氢酶1的活性与其内源活性相比减弱或失活,(vi)以使得丙酮酸脱羧酶1的活性与其内源活性相比减弱或失活,和/或(vii)以使得D-乳酸脱氢酶1的活性与其内源活性相比减弱或失活。另外,本申请的微生物可以经进一步修饰(i)以使得乙醇脱氢酶1(ADH1)的活性与其内源活性相比失活;(ii)以使得丙酮酸脱羧酶1(PDC1)的活性与其内源活性相比失活;和(iii)以使得D-乳酸脱氢酶1(DLD1)的活性与其内源活性相比失活。如本文所使用的,术语“丙酮酸脱羧酶(PDC)”指将丙酮酸转化为乙醛和二氧化碳(CO2)的酶,该丙酮酸脱羧酶可以与催化丙酮酸的脱羧的酶互换使用。丙酮酸脱羧酶是在酵母中特别是酵母属中发生的厌氧条件下的发酵过程中涉及的酶,并且是通过发酵生产乙醇的酶。通常,在酵母属中,PDC以三种不同形式的同工酶存在,即PDC1、PDC5和PDC6。PDC的蛋白质和基因序列可由已知的数据库如NCBI的GenBank获得,但不限于此。具体而言,关于该酶,PDC1可以是由氨基酸序列SEQIDNO:39表示的蛋白质,PDC5可以是由氨基酸序列SEQIDNO:41表示的蛋白质,PDC6可以是由氨基酸序列SEQIDNO:43表示的蛋白质,但可以无限制地包括具有PDC活性的任何氨基酸序列。另外,编码PDC1、PDC5和PDC6的基因可以分别由例如核苷酸序列SEQIDNO:40、42和44来具体表示,但可以无限制地包括可以编码该酶的任何核苷酸序列。如本文所使用的,术语“ATP-柠檬酸裂解酶(ACL,EC2.3.3.8)”指将柠檬酸转化为草酰乙酸(OAA)和乙酰辅酶A的酶,且已知存在于高等生物和一些酵母中(ATP-citratelyase:Amini-review,BiochemicalandBiophysicalResearchCommunications,422,(2012),1-2)。反应方案如下所示:柠檬酸+ATP+CoA+H2O→OAA+乙酰辅酶A+A+Pi乙酰辅酶A是微生物生长所必需的酶,其重要性近来已在各种参考文献中被强调。作为代表性的实例,有一篇报道,其为关于通过非天然途径提供胞质乙酰辅酶A来提高能够生产1,3-丁二醇(1,3-BDO)的真核生物的生产力的研究(国际专利公开号WO2013/036764)。在这点上,可以通过引入外源性ACL来提供对于其中PDC的活性被减弱或去除的菌株的生长所必需的乙酰辅酶A,由此使得微生物能够以独立于PDC活性的方式生长。蛋白质和基因序列可以由已知的数据库如NCBI的GenBank等获得,但不限于此。具体而言,ATP-柠檬酸裂解酶可以具有氨基酸序列SEQIDNO:29,但可以无限制地包括具有酶活性的任何蛋白质序列。另外,编码ACL的基因可以由核苷酸序列SEQIDNO:30来具体表示,但可以无限制地包括编码该酶的任何序列。如本文所使用的,术语“丙酮酸生物合成途径”指可以在酵母属微生物中提供丙酮酸的生物合成途径,其可以是从OAA到丙酮酸的供给路线。具体实例示于图1和2中。在一个示例性的实施方案中,丙酮酸生物合成途径可以通过修饰磷酸烯醇式丙酮酸羧激酶1(PCK1)的活性、或丙酮酸激酶2(PYK2)的活性、或这两种酶的活性以增强它们与其内源活性相比的活性来进行。或者,丙酮酸生物合成途径可以通过修饰苹果酸脱氢酶2(MDH2)的活性、或胞质苹果酸酶1(胞质MAE1)的活性、或两种酶的活性以增强它们与其内源活性相比的活性来进行。如本文所使用的,术语“磷酸烯醇式丙酮酸羧激酶1(PCK1)”指催化OAA向磷酸烯醇式丙酮酸(PEP)转化的酶。PCK1是在酵母微生物中将OAA转化为PEP的糖异生所必需的酶,并且已知其表达在葡萄糖的存在下被抑制(Differentialpost-transcriptionalregulationofyeastmRNAsinresponsetohighandlowglucoseconcentrations.MolMicrobiol35(3):553-65(2000))。如本文所使用的,术语“丙酮酸激酶2(PYK2)”指通过从PEP向ADP递送磷酸基团来催化丙酮酸和ATP的产生的酶。PYK2是酵母微生物中糖酵解的最后一步中的酶,也已知其表达在葡萄糖的存在下被抑制(Characterizationofaglucose-repressedpyruvatekinase(Pyk2p)inSaccharomycescerevisiaethatiscatalyticallyinsensitivetofructose-1,6-bisphosphate,JBacteriol.1997May;179(9):2987-93)。每一种蛋白质和基因序列可以由已知的数据库如NCBI的GenBank等获得,但不限于此。PCK1可以具有氨基酸序列SEQIDNO:31,但可以无限制地包括具有该酶活性的任何蛋白质序列。另外,编码PCK1的基因可以由核苷酸序列SEQIDNO:32来具体表示,但可以无限制地包括编码该酶的任何序列。PYK2可以具有氨基酸序列SEQIDNO:33,但可以无限制地包括具有该酶活性的任何蛋白质序列。另外,编码PYK2的基因可以由核苷酸序列SEQIDNO:34来表示,但可以无限制地包括编码该酶的任何序列。如本文所使用的,术语“苹果酸脱氢酶2(MDH2)”指将OAA转化为苹果酸的可逆酶。MDH2是一种原本位于胞质中的酶。如本文所使用的,术语“胞质苹果酸酶(MAE1)”指通过从MAE1去除线粒体靶向序列来修饰以位于胞质中的酶,该酶是用丙酮酸替代苹果酸的酶。MAE1酶是原本位于线粒体中的蛋白质,它在线粒体中将三羧酸(TCA)循环中的中间体物质苹果酸转化为丙酮酸(MetabolicEngineering,6(2004),352-363)。每一种蛋白质和基因序列可以由已知的数据库如NCBI的GenBank等获得,但不限于此。MDH2可以具有氨基酸序列SEQIDNO:35,但可以无限制地包括具有该酶活性的任何蛋白质序列。另外,作为具体的实例,编码MDH2的基因可以由核苷酸序列SEQIDNO:36表示,但可以无限制地包括可以编码该酶的任何序列。MAE1可具有氨基酸序列SEQIDNO:37,但可以无限制地包括具有该酶活性的任何蛋白质序列。另外,对于待存在于胞质中的MAE1,MAE1可以具有氨基酸序列SEQIDNO:37的序列,其中从开始到位置30的位置处的氨基酸残基被去除(即,为线粒体靶向序列的氨基酸序列SEQIDNO:51自其去除的序列),并且该序列由SEQIDNO:52表示。另外,在具体的实例中,编码MAE1的基因可以由核苷酸序列SEQIDNO:38表示,但可以无限制地包括可以编码该酶的任何序列。如本文所使用的,术语“乳酸脱氢酶(LDH)”指可以催化乳酸向丙酮酸及反过来丙酮酸向乳酸转化的酶,并且蛋白质和基因序列可以由已知的数据库如NCBI的GenBank等获得,但不限于此。LDH可以具有氨基酸序列SEQIDNO:49,但可以无限制地包括具有酶活性的任何蛋白质序列。另外,编码LDH的基因可以由核苷酸序列SEQIDNO:50表示,但可以无限制地包括可以编码该酶的任何序列。除了由SEQIDNO表示的氨基酸序列外,上述每种酶可以无限制地包括与上面列出的每一种氨基酸序列具有70%或更高、特别地80%或更高、更特别地90%或更高、甚至更特别地95%或更高、还甚至更特别地98%或更高、又甚至更特别地99%或更高的同源性的任何氨基酸序列,只要该酶实际上表现出与每一种所述的酶相同或相应的效果即可。另外,显而易见的是,具有上述同源性并具有与每一种酶相应的效果的任何经修饰的酶均可以属于本申请的范围,但所述酶可以具有部分缺失、修饰、替代或添加的氨基酸序列。另外,除了由SEQIDNO表示的核苷酸序列外,编码每一种酶的基因还可以无限制地包括与上面列出的每一种核苷酸序列具有80%或更高、特别地90%或更高、更特别地95%或更高、甚至更特别地98%或更高、还甚至更特别地99%或更高的同源性的编码这些酶的任何基因序列,只要该序列编码与每一种所述的酶具有基本上相同或相应的效果的酶即可。另外,显而易见的是,具有上述同源性的任何核苷酸序列均可以属于本申请的范围,但所述序列可以在其中具有部分缺失、修饰、替代或添加。如本文所使用的,术语“同源性”指两个多核苷酸或多肽部分之间的同一性的百分数。从一个部分到另一个部分的序列对应可以通过本领域已知的技术确定。例如,可以通过使用易于获得并且能够比对序列信息的计算机程序(例如,BLAST2.0)直接比对两个多核苷酸分子或两个多肽分子上的序列信息(例如,参数如得分、同一性和相似性)来确定同源性。另外,可以通过在同源区中形成稳定双链的条件下杂交多核苷酸并由单链特异性核酸酶消化该杂交链以确定被消化片段的大小来确定同源性。如本文所使用的,术语“内源活性”指其中微生物具有酶的自然状态或相应酶的修饰前的酶活化水平的情况。如本文所使用的,术语“修饰酶的活性以与其内源活性相比失活”指与天然菌株或修饰前的菌株相比,编码酶的基因根本不表达,或者即便在基因被表达时,也没有活性或活性降低。上面的降低是一种概念,其包括其中由于编码酶的基因的修饰等而使酶自身的活性与微生物原本具有的内源活性相比降低的情况,其中细胞内酶活性的整体水平与野生型菌株或修饰前菌株相比为较低的情况,以及其组合。酶的失活可以通过本领域已知的各种方法实现。所述方法的实例可以包括用突变以降低酶活性的基因替代染色体上编码酶的基因的方法,包括其中去除酶活性的情况;在染色体上编码酶的基因的表达调控序列中引入修饰的方法;用具有弱活性或无活性的序列替代编码酶的基因的表达调控序列的方法;删除染色体上编码酶的基因的全部或一部分的方法;引入反义寡核苷酸(例如,反义RNA)的方法,所述反义寡核苷酸与染色体上基因的转录物互补结合,从而抑制从mRNA向酶的翻译;将与SD序列互补的序列人工并入到编码酶的基因的SD序列的上游中,形成二级结构从而使核糖体不能与其连接的方法;向开放阅读框(ORF)的3’端并入启动子以诱导逆转录(逆转录工程(RTE))的方法等,以及其组合,但不限于此。具体而言,删除编码酶的基因的全部或一部分的方法可以通过使用用于菌株内染色体插入的载体以核酸序列中具有部分缺失的多核苷酸或标记基因替代染色体内编码内源性靶蛋白的多核苷酸来进行。在删除基因的一部分或全部的方法的示例性实施方案中,可以使用通过同源重组来删除基因的方法。如本文所使用的,术语“一部分”可以根据多核苷酸的种类变化,并且其可以具体指1至300,更具体地1至100,甚至更具体地1至50,但并不特别限于此。如本文所使用的,术语“同源重组”指通过在具有相互同源性的遗传链基因座处交换而发生的遗传重组。具体而言,表达调控序列可以通过在表达调控序列的核酸序列中以缺失、插入、非保守性或保守性替代或其组合来诱导表达调控序列的修饰而被修饰;或通过用较弱的启动子替代而被修饰等。表达调控序列可以包括启动子、操纵基因序列、编码核糖体结合区的序列和控制转录和翻译的终止的序列。此外,染色体上的基因序列可以通过在基因序列中以缺失、插入、非保守性或保守性替代或其组合来在序列中诱导修饰而被修饰以降低酶活性;或者通过用经改善而具有较弱活性的基因序列或经改善而不具有活性的基因序列替代。如本文所使用的,术语“与其内源活性相比活性增强”指通过修饰蛋白质以与蛋白质在其自然状态下所具有的活性相比提高细胞内活性来提高微生物中蛋白质(或酶)的细胞内活性。“增强”可以包括由于蛋白质(或酶)自身的活性提高而得出比原始功能更高的效果,并且其可以通过选自以下的至少一种方法进行:增加编码蛋白质(或酶)的多核苷酸的拷贝数的方法、在编码蛋白质(或酶)的基因的调控序列中引入修饰的方法、用具有强活性的序列替代染色体上编码蛋白质(或酶)的基因的调控序列的方法、用突变基因替代编码蛋白质(或酶)的基因以提高蛋白质(或酶)的活性的方法、和在染色体上编码蛋白质(或酶)的基因中引入修饰以增强蛋白质(或酶)的活性的方法,但可以无限制地包括可以与其内源活性相比增强蛋白质(或酶)的活性或增强引入的活性的任何已知方法。如本文所使用的,术语“引入蛋白质(或酶)的活性”指向不具有特定蛋白质(或酶)的活性的微生物提供特定蛋白质(或酶)的活性;或通过在向不具有特定蛋白质(或酶)的活性的微生物提供特定蛋白质(或酶)的活性之后修饰该微生物以进一步改善所述蛋白质(或酶)的细胞内活性来提高微生物中特定蛋白质(或酶)的细胞内活性。“引入蛋白质(或酶)的活性”可以本领域已知的各种方法进行,例如:向染色体中插入包含编码蛋白质(或酶)的核苷酸序列的多核苷酸的方法;通过方法如经由向载体系统中引入来向微生物引入多核苷酸从而增加多核苷酸的拷贝数的方法;向编码蛋白质(或酶)的核苷酸序列的上游区中引入能够表现出提高的活性的启动子或引入在启动子中具有修饰的蛋白质(或酶)的方法;引入编码蛋白质(或酶)的核苷酸变异序列的方法等,但可以无限制地包括可以引入蛋白质(或酶)的活性的任何已知方法。在上文中,多核苷酸的拷贝数的增加可以以其中有效连接多核苷酸到载体的形式或通过向宿主细胞的染色体中插入多核苷酸来实现,但方法并不特别限于此。具体而言,多核苷酸的拷贝数的增加可以通过引入载体来实现,无论宿主细胞如何,该载体都可以复制和起作用并且编码本申请的蛋白质的多核苷酸有效连接到其上;或者可以通过向宿主细胞中引入载体来实现,该载体可以向宿主细胞的染色体中插入多核苷酸并且多核苷酸有效连接到其上。载体是一种包含编码目标肽的多核苷酸序列的DNA构建体,其有效连接到合适的调控序列以使目标肽能够在宿主细胞中表达。调控序列包括能够启动转录的启动子、用于调控转录的任何操纵基因序列、编码适当的mRNA核糖体结合域的序列、以及调控转录和翻译的终止的序列。在转化到合适的宿主细胞中后,载体可以被复制或起作用而无论宿主基因组如何,或者可以被整合到宿主基因组自身中。对于酵母表达载体,可以使用整合型酵母质粒(YIp)和染色体外质粒载体二者。染色体外质粒载体可以包括附加型酵母质粒(YEp)、复制型酵母质粒(YRp)和酵母着丝粒质粒(YCp)。另外,根据本申请,也可以使用人造酵母染色体(YAC)作为表达载体。例如,在本申请中待使用的载体可以包括pESC-HIS、pESC-LEU、pESC-TRP、pESC-URA、GatewaypYES-DEST52、pAO815、pGAPZA、pGAPZB、pGAPZC、pGAPαA、pGAPαB、pGAPαC、pPIC3.5K、pPIC6A、pPIC6B、pPIC6C、pPIC6αA、pPIC6αB、pPIC6αC、pPIC9K、pYC2/CT、pYD1YeastDisplayVector、pYES2、pYES2/CT、pYES2/NTA、pYES2/NTB、pYES2/NTC、pYES2/CT、pYES2.1、pYES-DEST52、pTEF1/Zeo、pFLD1、PichiaPinkTM、p427-TEF、p417-CYC、pGAL-MF、p427-TEF、p417-CYC、PTEF-MF、pBY011、pSGP47、pSGP46、pSGP36、pSGP40、ZM552、pAG303GAL-ccdB、pAG414GAL-ccdB、pAS404、pBridge、pGAD-GH、pGADT7、pGBKT7、pHIS-2、pOBD2、pRS408、pRS410、pRS418、pRS420、pRS428、酵母微米A形式、pRS403、pRS404、pRS405、pRS406、pYJ403、pYJ404、pYJ405和pYJ406,但不限于此。更具体而言,酵母载体可以是包含复制起点(ori)的酵母复制质粒和可以在大肠杆菌(E.coli)中增殖和选择的抗生素抗性盒。通常,表达载体可以包括启动子-基因-转录终止序列的表达构建体。例如,当宿主细胞为酵母时,可以用于表达构建体中的启动子可以包括TEF1启动子、TEF2启动子、GAL10启动子、GAL1启动子、ADH1启动子、ADH2启动子、PHO5启动子、GAL1-10启动子、TDH3启动子(GPD启动子)、TDH2启动子、TDH1启动子、PGK1启动子、PYK2启动子、ENO1启动子、ENO2启动子和TPI1启动子,但不限于此。可以用于表达构建体中的转录终止序列可以包括ADH1终止子、CYC1终止子、GAL10终止子、PGK1终止子、PHO5终止子、ENO1终止子、ENO2终止子和TPI1终止子,但不限于此。另外,编码内源性靶蛋白的多核苷酸可以由用于在宿主细胞内插入染色体的载体用染色体内经修饰的多核苷酸置换。或者,编码待引入到染色体中的外源靶蛋白的多核苷酸可以用经修饰的多核苷酸置换。向染色体中插入多核苷酸可以使用本领域中的任何已知的方法进行,例如通过同源重组。由于本申请的载体可以经由同源重组插入到染色体中,故可以进一步包括用于确认插入到染色体中的选择标记。选择标记用于选择转化的细胞,即确认目标多核苷酸是否已被插入,并可以使用能够提供可选择的表型如耐药性、营养需求、对细胞毒性剂的抗性和表面蛋白的表达的标记。在用选择剂处理的情况下,只有能够表达选择标记的细胞才能存活或表达其他表型性状,从而可以选择转化的细胞。如本文所使用的,术语“转化”指向宿主细胞中引入包含编码靶蛋白的多核苷酸的载体从而允许由蛋白质编码的多核苷酸在宿主细胞中的表达的过程。对于转化的多核苷酸,其是否插入到宿主细胞的染色体中以及是位于其中还是位于染色体外部都不重要,只要其可以在宿主细胞中表达即可。另外,多核苷酸包括编码靶蛋白的DNA和RNA。多核苷酸可以以任何形式插入,只要其可以被引入到宿主细胞中并在其中表达即可。例如,多核苷酸可以以表达盒的形式引入到宿主细胞中,表达盒是包括自我表达所需的所有必需元件的基因构建体。表达盒通常可以包括可操作地连接到多核苷酸的启动子、转录终止信号、核糖体结合域和翻译终止信号。表达盒可以是能够自我复制的表达载体的形式。另外,可以将多核苷酸原样引入宿主细胞中并有效连接到其在宿主细胞中表达所必需的序列。另外,如本文所使用的,术语“可操作地连接”指在启动和介导编码本申请的靶蛋白的多核苷酸的转录的启动子序列与上述靶基因序列之间的功能连接。转化本申请的载体的方法可以包括可以向细胞中引入核酸的任何方法,并且转化可以通过根据宿主细胞选择本领域已知的合适技术来进行。例如,方法可以包括电穿孔、磷酸钙(CaPO4)沉淀、氯化钙(CaCl2)沉淀、显微注射、聚乙二醇(PEG)法、DEAE-葡聚糖法、阳离子脂质体钙和乙酸锂/DMSO法等,但不限于此。具体而言,待使用的宿主细胞应该具有高的DNA引入效率和引入DNA的高表达效率,并且就本申请的目的而言,宿主细胞可以是酵母属微生物。然后,虽然并不特别限于此,但为了增加多核苷酸的表达而在表达调控序列中引入修饰可以通过经由缺失、插入、保守性替代或非保守性替代或其组合诱导核酸序列中的修饰来进行,以进一步增强表达调控序列的活性;或通过用具有增强活性的核酸序列置换多核苷酸序列。表达调控序列可以包括启动子、操纵基因序列、编码核糖体结合域的序列及调控转录和翻译的终止的序列等,但并不特别限于此。可以将强的外源启动子而不是原启动子连接到多核苷酸的表达单元的上游区。通常,蛋白质的活性的引入或增强可以将相应蛋白质的活性或浓度相对于野生型蛋白质的活性或浓度或者微生物菌株中的活性或浓度提高至少10%、25%、50%、75%、100%、150%、200%、300%、400%或500%至最大1000%或2000%。在另一个方面,本申请提供了一种生产乳酸的方法,其包括:a)培养新型的产乳酸酵母属微生物,其中所述微生物经修饰以使丙酮酸脱羧酶(PDC)的活性与其内源活性相比失活,以引入ATP-柠檬酸裂解酶(ACL)的活性,并以增强与其内源生物合成途径相比在培养基中的丙酮酸生物合成途径;和b)从培养的微生物和培养物中回收乳酸。产乳酸酵母属微生物与上文所述相同。如本文所使用的,术语“培养”指在适当人工调节的环境中生长微生物。在本申请中,使用酵母属微生物的培养可以通过本领域众所周知的合适方法进行。具体而言,培养可以分批过程、补料分批过程或重复补料分批过程连续地进行,但不限于此。用于培养本申请的微生物的培养基和其他培养条件不受特别限制,但可以使用用于酵母属微生物的常规培养的任何培养基。具体而言,本申请的微生物可以在含有适当碳源、氮源、磷源、无机化合物、氨基酸和/或维生素等的常规培养基中、在需氧条件下、同时调节温度、pH等进行培养。作为碳源的一个实例,可以使用蔗糖或葡萄糖,还可使用含有大量蔗糖的糖蜜作为碳源,并且可以使用适量的其他各种碳源。氮源的实例可以包括有机氮源如蛋白胨、酵母提取物、肉汁、麦芽提取物、玉米浆和大豆粉;和无机氮源如尿素、硫酸铵、氯化铵、磷酸铵、碳酸铵和硝酸铵。这些氮源可以单独使用或组合使用。在上述培养基中,可以含有磷酸二氢钾、磷酸氢二钾和相应的含钠盐作为磷源。另外,也可含有金属盐如硫酸镁或硫酸铁。此外,可以含有氨基酸、维生素和合适的前体。这些培养基或前体可以在分批培养过程或连续培养过程中添加到培养物中。在培养的过程中,可以通过以适宜的方式向培养物添加化合物如氢氧化铵、氢氧化钾、氨水、磷酸和硫酸来调节培养物的pH。另外,在培养的过程中,可以添加消泡剂如脂肪酸多元醇酯来防止泡沫生成。另外,为了维持培养物的好氧状态,可以向培养物中注入氧气或含氧气体,而为了维持培养物的厌氧和微需氧状态,可以注入氮气、氢气或二氧化碳气体而不注入空气。培养物温度通常可以为20℃至40℃,特别地为25℃至35℃,更特别地为30℃,但可以根据所期望目的无限制地改变。另外,培养可以继续直至可以获得期望量的产物,具体而言,为10小时至100小时,但不限于此。本申请的生产乳酸的方法可以包括从培养的微生物或培养物中回收乳酸。从微生物或培养物中回收乳酸的方法可以使用本领域已知的合适方法进行,例如,离心、过滤、阴离子交换色谱、结晶、HPLC等,但不限于此。回收可以包括纯化过程。实施例下文将结合示例性实施方案详细地描述本申请。然而,本文公开的示例性实施方案仅出于说明目的而不应理解为限制本申请的范围。实施例1:产乳酸菌株的制备为了制备本申请中待用的代表性产乳酸菌株,使由Euroscarf获得的野生型酵母菌株中的代表性酵母菌株——酿酒酵母CEN.PK2-1D经受一系列遗传操作。具体而言,删除乙醇脱氢酶1(ADH1)和丙酮酸脱羧酶1(PDC1)以使产生醇的发酵最小化,而为了阻断乳酸分解的途径,使用缺失D-乳酸脱氢酶1(DLD1)的菌株作为本申请的基础菌株。DLD1不是直接影响生长改善的因素,但作为D-型乳酸的脱氢酶的DLD1已知是使用NAD+将乳酸转化为丙酮酸的主要酶。相应地,基于缺失了为乳酸消耗酶的DLD1基因的菌来制备后续的菌株,并比较乳酸生产力。在本申请中,使用通用分子克隆方法进行遗传操作。首先,基于参考文献(LeeTH等人,J.Microbiol.Biotechnol.(2006),16(6),979-982)上的公开内容,使用pWAL100和pWBR100质粒进行酶的ADH1和PDC1基因的删除实验。通过聚合酶链反应(PCR),使用对应于每一个插入物(SEQIDNO:1至SEQIDNO:8)的引物,制备合并到每一个质粒中的每一个插入物。使用野生型酵母菌株的基因组DNA作为模板进行PCR。对于ADH1的删除,使用SEQIDNO:1和SEQIDNO:2的引物进行PCR,并使用限制性酶BamHI和NcoI将所得物克隆到pWAL100中。使用SEQIDNO:3和SEQIDNO:4的引物进行额外的PCR,并使用限制性酶BamHI和NcoI将所得物克隆到pWBR100中。通过在95℃下变性5分钟、在53℃下退火1分钟和在72℃下聚合1分30秒来进行PCR。对于PDC1的删除,使用SEQIDNO:5和SEQIDNO:6的引物进行PCR,并使用限制性酶BamHI和NcoI将所得物克隆到pWAL100中。使用SEQIDNO:7和SEQIDNO:8的引物进行额外的PCR,并使用限制性酶BamHI和NcoI将所得物克隆到pWBR100中。通过在95℃下变性5分钟、在53℃下退火1分钟和在72℃下聚合1分30秒来进行PCR。另外,对于DLD1基因的删除,通过经由双交换的引入来删除HIS3标记基因。其中使用的DNA片段通过使用野生型酵母菌株的基因组DNA连同SEQIDNO:9和SEQIDNO:10的引物进行PCR来获得。通过在95℃下变性5分钟、在53℃下退火1分钟和在72℃下聚合1分30秒来进行PCR。基因操作中使用的引物汇总于下表1中。[表1]用于制备乳酸生产用基础菌株的引物序列基于缺失三个基因(ADH1、PDC1和DLD1)的菌株,引入用于乳酸生产的D-乳酸脱氢酶(D-LDH)。将源自植物乳杆菌(Lb.plantarum)的ldhD基因的5’端和3’端分别插入到p413TEF1载体中,使得ldhD基因可以被包含在TEF1启动子和源自酿酒酵母的CYC1终止子之间,其中通过用SaxI/PvuII双消化来制备插入物。在用p-δ-neo载体的BamHI/NotI双消化的DNA片段中使用绿豆核酸酶使载体平头化,并用SacI再次处理,从而产生具有SacI粘端和BamHI平端的载体部分。将由此获得的载体和插入物连接以完成pTL573载体并命名为pTL573载体。pTL573质粒含有源自植物乳杆菌的ldhD基因,并设计为使得可以将多个拷贝随机插入到δ-序列中,该序列是酿酒酵母CEN.PK2-1Dpdc1adh1dld1菌株的反转录转座因子的区域的一部分。为了相应基因的多重插入,用SacI消化pTL573质粒以制备可以在δ-序列中诱导单交换的DNA片段。通过转染将所得物引入到亲本菌株中,并在最大浓度为5mg/mLG418的YPD(1%的酵母提取物、2%的细菌蛋白胨和2%的葡萄糖)培养基中获得众多菌落。证实由此获得的菌株最终插入了多个源自植物乳杆菌的D-LDH以提供D-型乳酸生产力并将该菌株命名为CC02-0064。实施例2:具有降低的滴度或失活的PDC的菌株的制备通过制备在CC02-0064菌株中缺失了PDC5即一种PDC同工酶的菌株及在CC02-0064菌株中缺失了PDC5和PDC6二者的菌株来制备具有降低的滴度或PDC失活的菌株,CC02-0064菌株为实施例1中制备的基础菌株。具体而言,对于酵母的基因删除,使用pWAL100和pWBR100质粒(J.Microbiol.Biotechnol.,(2006)16(6),979-982)。对于PDC5的删除,使用SEQIDNO:11和SEQIDNO:12的引物进行PCR,并使用限制性酶BamHI和NotI将所得物克隆到pWAL100中。使用SEQIDNO:13和SEQIDNO:14的引物进行额外的PCR,并使用限制性酶SpeI和NcoI将所得物克隆到pWBR100中。通过在95℃下变性5分钟、在53℃下退火1分钟和在72℃下聚合1分30秒来进行PCR。对于PDC6的删除,使用SEQIDNO:15和SEQIDNO:16的引物进行PCR,并使用限制性酶BamHI和NotI将所得物克隆到pWAL100中。使用SEQIDNO:17和SEQIDNO:18的引物进行额外的PCR,并使用限制性酶SpeI和NcoI将所得物克隆到pWBR100中。通过在95℃下变性5分钟、在53℃下退火1分钟和在72℃下聚合1分30秒来进行PCR。[表2]用于删除PDC同工酶的引物序列上面制得的新型菌株分别命名为CC02-0256和CC02-0553,新型菌株的遗传性状汇总于下表3中。[表3]具有降低或失活的PDC活性的菌株实施例3:具有降低或失活的PDC活性的菌株的乳酸生产力评价用于菌株评价的培养基为合成复合培养基(SC)。为了制备该培养基,根据制造商的方案将氨基酸缺陷型混合物(aminoaciddropoutmix,Sigma)与用作碱的0.67%无氨基酸酵母氮源混合,并根据需要加入其中不包含的氨基酸。添加亮氨酸至380mg/L的浓度,添加尿嘧啶、色氨酸和组氨酸至分别为76mg/L的浓度并向其添加葡萄糖(8%)作为碳源和1%CaCO3作为中和剂。使用由此制得的培养基评价酵母菌株的乳酸发酵。作为评价菌株的乳酸发酵能力的条件,将为评价乳酸发酵所制得的培养基以每个烧瓶25mL的量等分并接种每一种酵母菌株,在30℃下需氧培养48小时,并通过HPLC分析发酵液中存在的乳酸量。实验结果汇总于下表4中。[表4]具有降低或失活的PDC活性的菌株的发酵结果比较结果是如由上表4可确认的,随着PDC活性降低,产率增加但生产力降低。实施例4:基于PDC-失活菌株引入ATP-柠檬酸裂解酶(ACL)及制备具有增强的磷酸烯醇式丙酮酸羧激酶1(PCK1)和丙酮酸激酶2(PYK2)活性的菌株(1)制备用于向产乳酸菌株中引入外源ACL的载体制备用于引入外源ACL酶并同时过表达PCK1和PYK2的重组载体,PCK1和PYK2为丙酮酸生物合成的途径之一。对于外源ACL,使用源自哺乳动物小家鼠(Musmusculus)的基因,并通过NCBI(登录号NP_001186225)确认相应的基因。具体而言,使用氨基酸序列SEQIDNO:29(或氨基酸序列SEQIDNO:30)合成基因,使用基于pRS415的GPD启动子制备载体,pRS415为一种用于酵母的基因表达载体,并制备插入了该基因的载体,命名为p415GPDpro-ACL。(2)制备具有增强的PCK1和PYK2的载体以增强丙酮酸生物合成途径制备用于同时过表达PCK1和PYK2以增强丙酮酸生物合成途径的重组载体。PYK2是存在于酵母微生物中的基因并可以由SEQIDNO:33表示。使用酿酒酵母的基因组DNA作为模板连同SEQIDNO:19和20的引物进行PCR,由此获得PYK2基因的片段。通过在95℃下变性5分钟、在53℃下退火1分钟和在72℃下聚合1分30秒来进行PCR。使用这些基因片段和源自pRS416的酵母表达载体内的限制性酶,即SpeI、XhoI进行克隆,并使用TEF1启动子进行过表达。相应的重组载体命名为pRS416-TEF1pro-PYK2。PYK1也是存在于酵母微生物中的基因,并可以由SEQIDNO:31表示。使用酿酒酵母的基因组DNA作为模板连同SEQIDNO:23和24的引物进行PCR,由此获得PYK1基因的片段。通过在95℃下变性5分钟、在53℃下退火1分钟和在72℃下聚合1分30秒来进行PCR。在与使用酿酒酵母的基因组DNA作为模板连同SEQIDNO:21和22的引物获得PCK1片段的相同条件下进行PCR,使得可以使用TEF2启动子表达PYK1,并获得TEF2启动子的片段。然后,为了在单个重组载体中同时表达PCK1和PYK2,用XhoI消化上面制得的pRS416-TEF1pro-PYK2重组载体,同时,使用In-Fusion克隆试剂盒(Clontech)克隆TEF2启动子的片段和PCK1片段。最后,得到可以分别用TEF2启动子和TEF1启动子过表达PCK1和PYK2的单个重组载体,并将该载体命名为pRS416-TEF1pro-PYK2-TEF2pro-PCK1。用于过表达PCK1和PYK2的载体的制备中使用的引物汇总于下表5中。[表5]用于过表达PCK1和PYK2的引物(3)向具有失活的PDC的产乳酸菌株中引入外源ACL和制备具有增强的PCK1和PYK2活性的菌株基于实施例2中制备的CC02-0553菌株,引入实施例4-(1)中制备的外源ACL,并通过转染插入实施例4-(2)中制备的用于同时过表达PCK1/PYK2的载体。进行转染的方法包括用含有0.1M乙酸锂、0.01MTris-HCl和0.001MEDTA的溶液(下文称为LiAc/TE缓冲液)处理在YPD(1%的酵母提取物、2%的细菌蛋白胨和2%的葡萄糖)培养基中培养的CC02-0553菌株18小时,并与含有40%PEG的LiAc/TE缓冲液一同在42℃下热处理15分钟以插入重组载体。由此制备的菌株分别命名为CC02-0652和CC02-0765,遗传性状汇总于下表6中。[表6]基于具有失活的PDC效力的菌株及具有增强的PCK1和PYK2活性的菌株引入ACL实施例5:基于失活的PDC引入ACL及对在具有增强的PCK1和PYK2活性的菌株中的发酵的评价为了评价ACL-PCK1-PYK2增强菌株,以与实施例3中相同的方式,在实施例4-(3)中制备的具有失活的PDC效力的菌株中评价乳酸发酵能力。结果汇总于下表7中。[表7]基于具有失活的PDC滴度的菌株引入ACL及具有增强的PCK1和PYK2活性的菌株的评价菌株OD(600nm)葡萄糖消耗(g/L)乳酸(g/L)产率(%)生产力(g/L/h)CC02-05532.634.520.860.20.43CC02-06525.056.031.662.50.66CC02-07656.169.145.666.00.95结果是如上表7中可确认的,其中引入了外源ACL并且其中增强了PCK1和PYK2活性的菌株表现出OD600值增加了130%,该OD600值代表相对于PDC-失活菌株的细菌生长;同期葡萄糖消耗的量增加了100%;并且乳酸发酵产率提高了10%。另外,菌株最终显示出乳酸生产力提高了120%。基于CC02-0652菌株的发酵结果,确认了外源ACL的引入可以由于新的生产途径产生乙酰辅酶A而增强酵母微生物的生长。另外确认了外源ACL的引入不仅可以增强生长,而且可以提高生产力。此外,根据CC02-0765菌株的发酵结果,确认了通过丙酮酸生物合成的增强,可以进一步提高乳酸发酵的生产力,并从而确认了通过本申请的采用由新的途径产生乙酰辅酶A并增强丙酮酸生物合成的策略生产乳酸的方法不同于现有技术,是一种不仅可以提高乳酸发酵产率和增强给定微生物的生长,而且可以提高乳酸发酵生产力的方法。因此,在2014年11月28日将CC02-0765菌株保藏在韩国微生物培养中心(KCCM),按布达佩斯条约的保藏号为KCCM11616P。实施例6:基于PDC-失活的菌株引入ACL及制备具有增强的苹果酸脱氢酶2(MDH2)和胞质苹果酸酶1(MAE1)活性的菌株基于实施例5的结果,确认了由新的途径产生乙酰辅酶A并增强丙酮酸生物合成的策略是提高乳酸发酵产率、增强具有生产力的微生物的生长和提高乳酸发酵生产力的有效方法,因此,本发明人试图确认使用其他基因来增强丙酮酸生物合成是否可以具有相似效果。(1)制备具有增强的MDH2和胞质MAE1活性的载体由于通过外源ACL的引入产生的OAA可以通过不同的途径生物合成为丙酮酸,故本发明人试图过表达MDH2并过表达MAE1,MDH2原本位于胞质中,MAE1是位于线粒体中的酶,通过改变其位置进入胞质中。为此,制备重组载体。MDH2是存在于酵母微生物中的基因,并可以由SEQIDNO:35的氨基酸序列表示。通过使用酿酒酵母的基因组DNA作为模板连同SEQIDNO:25和26的引物进行的PCR获得MDH2基因的片段。通过在95℃下变性5分钟、在53℃下退火1分钟和在72℃下聚合1分钟来进行PCR。在用限制性酶SpeI和XhoI消化后,基于pRS414载体克隆由此获得的MDH2基因的片段,其中将MDH2基因设定为使用TEF1启动子过表达。由此获得的重组载体命名为pRS414-TEF1pro-MDH2。MAE1是原本存在于酵母微生物的线粒体中的基因,并可以由SEQIDNO:37的氨基酸序列表示。为了在胞质中表达MAE1基因,将MAE1基因克隆,排除由距MAE1基因的起始密码子90个核苷酸的序列组成的线粒体靶序列(由SEQIDNO:51的氨基酸序列表示)。使用酿酒酵母的基因组DNA作为模板连同SEQIDNO:27和28的引物进行PCR。通过在95℃下变性5分钟、在53℃下退火1分钟和在72℃下聚合2分钟来进行PCR。在用限制酶SpeI和XhoI消化后,基于pRS416载体克隆由此获得的PCR片段。SEQIDNO:27的引物以获得从位置91开始的核苷酸序列的方式制备,以从MAE1ORF起始密码子开始去除90个核苷酸的序列。另外,试图使用TEF1启动子过表达胞质MAE1。另外,将由此制得的重组载体命名为pRS416-TEF1pro-胞质MAE1。用于制备重组载体的引物,即pRS414-TEF1pro-MDH2和pRS416-TEF1pro-胞质MAE1,汇总于下表8中。[表8]用于过表达MDH2和胞质MAE1的引物(2)在具有失活的PDC的产乳酸菌株中制备具有增强的MDH2和胞质MAE1活性的菌株基于实施例2中制备的CC02-0553菌株,通过转染克隆实施例4-(1)中制备的引入了外源ACL的载体和实施例6-(1)中制备的MDH2过表达载体和胞质MAE1过表达载体。转染使用实施例4-(3)中所说明的方法进行。由此制得的菌株命名为CC02-0821,该菌株的遗传性状汇总于下表9中。[表9]在具有失活的PDC的菌株及具有增强的MDH2和胞质MAE1活性的菌株中引入ACL实施例7:基于具有失活的PDC的菌株引入ACL以及对在具有增强的MDH2和胞质MAE1活性的菌株中的发酵的评价为了评价ACL-MDH2-胞质MAE1-增强菌株,以与实施例2中相同的方式在实施例6中制备的具有失活的PDC的菌株中评价乳酸发酵能力。结果汇总于下表10中。[表10]基于具有失活的PDC的菌株评价ACL-MDH2-胞质MAE1-增强菌株的发酵能力如上表10中可确认的,如CC02-0765菌株的情况一样,其中通过MDH2和胞质MAE1的过表达增强丙酮酸生物合成的CC02-0821菌株的发酵能力评价结果也表现出OD600值增加了110%,该OD600值代表相对于其中去除了PDC活性的菌株的细菌生长;同期葡萄糖消耗的量增加了80%;并且乳酸发酵产率提高了6%。另外,菌株最终显示出乳酸生产力提高了90%。基于上面的结果,确认了不仅外源ACL的引入将改善酵母微生物中的乳酸生产,而且经由多种路线增强丙酮酸生物合成途径对于进一步提高乳酸生产力也是有效的。实施例8:在具有降低的PDC活性的菌株中引入ACL及制备具有增强的PCK1和PYK2活性的菌株、具有增强的MDH2和胞质MAE1活性的菌株从实施例的结果可以确定,其中去除了PDC滴度的菌株可以通过引入外源ACL和增强丙酮酸生物合成途径来提高乳酸发酵生产力。因此,本发明人试图确认是否能从其中降低了PDC滴度的菌株以及其中去除了PDC滴度的菌株获得相同的结果。以与实施例4-(3)中相同的方式,基于CC02-0256菌株插入实施例4中制备的重组载体,即pRS415-GPDpro-ACL和pRS416-TEF1pro-PYK2-TEF2p-PCK1,CC02-0256菌株为实施例2中制备的具有降低的PDC活性的菌株。由此制得的菌株分别命名为CC02-0819和CC02-0820。此外,以与实施例4-(3)中相同的方式,基于CC02-0256菌株插入实施例4中制备的重组载体,即pRS415-GPDpro-ACL和实施例6-(1)中制备的重组载体pRS414-TEF1pro-MDH2和pRS416-TEF1pro-胞质MAE1,CC02-0256为实施例2中制备的具有降低的PDC活性的菌株。由此制得的菌株命名为CC02-0831,这些菌株的遗传性状汇总于下表11中。[表11]基于具有降低的PDC活性的菌株引入ACL及制备具有增强的丙酮酸生物合成途径的菌株实施例9:在具有降低的PDC活性的菌株中引入ACL及对在具有增强的PCK1和PYK2活性的菌株中的发酵的评价以与实施例3中相同的方式评价实施例8中制备的CC02-0819和CC02-0820菌株连同为对照组的CC02-0256菌株的乳酸发酵能力。实验结果汇总于下表12中。[表12]基于具有降低的PDC活性的菌株引入ACL以及具有增强的PCK1和PYK2活性的菌株的发酵评价结果是如上表12中可确认的,与CC02-0553菌株不同,CC02-0256菌株未因增加外源ACL的引入和增强丙酮酸生物合成途径而显示出效果的显著增加,但乳酸发酵产率提高,具有生产力的微生物的细菌生长增加,乳酸发酵的生产力也提高。其中引入了外源ACL并且增强了PCK1和PYK2活性的CC02-0820菌株表现出OD600值增加了80%,该OD600值代表相对于具有降低的PDC滴度的菌株的细菌生长;同期葡萄糖消耗的量增加了40%;并且乳酸发酵产率提高了2%。另外,乳酸生产力最终提高了40%。与仅引入了外源ACL的CC02-0819菌株相比,CC02-0820菌株在OD值、同期葡萄糖消耗量和乳酸发酵产率方面均表现出提高,并且显示生产力提高了20%。实施例10:在具有降低的PDC活性的菌株中引入ACL及对在具有增强的MDH2和胞质MAE1活性的菌株中的发酵的对比评价以与实施例3中相同的方式评价实施例8中制备的CC02-0831菌株连同为对照组的CC02-0256及CC02-0819菌株。实验结果汇总于下表13中。[表13]基于具有降低的PDC滴度的菌株引入ACL及具有增强的MDH2和胞质MAE1活性的菌株的发酵评价结果是如上表13中可确认的,其中引入了外源ACL并且增强了MDH2和胞质MAE1活性的CC02-0831菌株相对于亲本菌株CC02-0256表现出OD600值增加了80%;同期葡萄糖消耗的量增加了30%;并且乳酸发酵产率提高了2%。另外,乳酸生产力最终提高了30%。与仅引入了外源ACL的CC02-0819菌株相比,CC02-0831菌株在OD值、同期葡萄糖消耗量和乳酸发酵产率方面均表现出提高,并且显示生产力提高了20%。上述结果支持了通过采用由新的途径产生乙酰辅酶A并增强丙酮酸生物合成的策略生产乳酸的方法不同于现有技术,是一种不仅可以提高乳酸发酵产率和增强具有生产力的微生物的生长,而且可以提高乳酸发酵生产力的方法。具体来说,结果表明,通过采用由于外源ACL的引入而由新的途径产生乙酰辅酶A并增强丙酮酸生物合成的策略制得的微生物不仅可以提高乳酸发酵产率和增强具有生产力的微生物的生长,而且可以提高乳酸发酵生产力,并从而可以作为优异的产乳酸微生物提供。从以上所述,本申请所属领域的技术人员应能够理解,本申请可以其他具体形式实施而不改变本申请的技术构思或本质特征。就这一点而言,本文公开的示例性实施方案仅出于示意的目的而不应理解为限制本申请的范围。相反,本申请旨在不仅涵盖这些示例性实施方案,而且涵盖可以包括在由所附的权利要求限定的本申请的精神和范围内的各种替代、修改、等价物和其他实施方案。PCT/RO/134表当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1