接枝半胱氨酸的聚硅氧烷嵌段共聚物及制备方法与应用与流程
本发明属于功能性高分子嵌段共聚物的制备领域,特别涉及一种接枝半胱氨酸的聚硅氧烷嵌段共聚物及制备方法与应用。
背景技术:
蛋白质在材料表面的吸附往往是材料表面生物污损的先决条件,抗蛋白吸附材料因而成为医用材料、海洋防污、膜分离等诸多领域的研究热点。两性离子聚合物在抗蛋白吸附的研究中通过形成具有阻隔作用的“水化层”而表现出了优异的性能,但是出于其本身良好的水溶性,在水溶液中两性离子聚合物常常难以在易受蛋白污染的疏水性表面聚集形成有效的稳定吸附层,因而在需要抗蛋白吸附的许多应用场合受到了限制。
因此,有必要研究得到一种能在水溶液中稳定吸附到表面从而发挥抗蛋白吸附作用的两性离子聚合物。
技术实现要素:
本发明的首要目的在于克服现有技术的缺点与不足,提供一种接枝半胱氨酸的聚硅氧烷嵌段共聚物的制备方法。
本发明的另一目的在于提供通过上述制备方法得到的接枝半胱氨酸的聚硅氧烷嵌段共聚物。
本发明的再一目的在于提供上述接枝半胱氨酸的聚硅氧烷嵌段共聚物的应用。
本发明的目的通过下述技术方案实现:一种接枝半胱氨酸的聚硅氧烷嵌段共聚物的制备方法,包括如下步骤:
(1)聚二甲基硅氧烷-聚甲基乙烯基硅氧烷嵌段共聚物的合成:在有机溶剂a中加入六甲基环三硅氧烷(d3),在惰性气体气氛中,以正丁基锂引发第一次聚合反应,得到溶液e;将d3和1,3,5-三乙烯基-1,3,5-三甲基环三硅氧烷(d3v)溶解在有机溶剂a中,得到溶液f,将溶液f和溶液e混合,进行第二次聚合反应;最后加入三甲基氯硅烷终止聚合反应;对反应得到的产物纯化,得到透明油状液体产物,即聚二甲基硅氧烷-b-聚甲基乙烯基硅氧烷嵌段共聚物(pdms-b-pvms);
(2)聚二甲基硅氧烷-聚硅氧烷接枝n-乙酰半胱氨酸嵌段共聚物的合成:将有机溶剂b、有机溶剂c、pdms-b-pvms、n-乙酰半胱氨酸和安息香二甲醚混合,得到澄清的溶液,用紫外光引发自由基加成反应,对反应得到的产物纯化,得到聚二甲基硅氧烷-b-聚硅氧烷接枝n-乙酰半胱氨酸嵌段共聚物【pdms-b-(pdms-g-acys)】;
(3)接枝半胱氨酸的聚硅氧烷嵌段共聚物的合成:在有机溶剂d中加入pdms-b-(pdms-g-acys)与盐酸,回流加热反应;对反应得到的产物纯化,得到接枝半胱氨酸的聚硅氧烷嵌段共聚物【pdms-b-(pdms-g-cys)】。
上文中所述的有机溶剂a、b、c、d是用于溶解反应物质,本身不参加反应。有机溶剂a、b、c、d可以是相同的物质,也可以是不同的物质。
上文中所述的惰性气氛优选为氮气气氛。
步骤(1)中所述的第一次聚合反应的条件优选为0~5℃聚合12h。
步骤(1)中所述的第二次聚合反应的条件优选为0~5℃聚合12h。
步骤(1)中所述的有机溶剂a优选为四氢呋喃和乙醚中的一种或两种。
步骤(1)中所述的溶液e中有机溶剂a的体积用量优选为相当于六甲基环三硅氧烷单体质量的2~4倍(ml:g)。
步骤(1)中所述的正丁基锂优选为先溶解在正己烷中,再加入到反应体系中;正丁基锂的用量与最终产物的摩尔量相当。
步骤(1)中所述的第一次聚合反应中d3的用量优选为按正丁基锂的摩尔量:d3的摩尔量=1:4.5~9配比。
步骤(1)中所述的第二次聚合反应中d3和d3v的用量优选为按与正丁基锂摩尔比:d3的摩尔量:d3v的摩尔量=1:18~22.5:18~22.5配比。
步骤(1)中所述的溶液f中有机溶剂a的体积用量优选为相当于六甲基环三硅氧烷单体、1,3,5-三乙烯基-1,3,5-三甲基环三硅氧烷单体的总质量(ml:g)。
步骤(1)中所述的纯化的步骤优选为:通过减压蒸馏除去溶剂以及未聚合的小分子原料,然后过滤除去副产物氯化锂粉末,得到纯化后的pdms-b-pvms。
步骤(2)中所述的pdms-b-pvms、n-乙酰半胱氨酸以及安息香二甲醚的配比优选为按照pdms-b-pvms中乙烯基的摩尔量:n-乙酰半胱氨酸的摩尔量:安息香二甲醚的摩尔量=1:(1.2~1.5):0.3配比。
步骤(2)中所述的紫外光优选为波长为350-365nm的紫外光;更优选为360nm的紫外光。
步骤(2)中所述的自由基加成反应的条件优选为室温下反应30~60min。所述的室温为15~30℃;更优选为25℃。
步骤(2)中所述的有机溶剂b优选为四氢呋喃、乙醚、三氯甲烷和二氯甲烷中的一种或至少两种。
步骤(2)中所述的有机溶剂c优选为甲醇、乙醇和异丙醇中的一种或至少两种。
步骤(2)中所述的有机溶剂b的质量用量优选为相当于pdms-b-pvms的总质量的5~6倍。
步骤(2)中所述的有机溶剂c的质量用量优选为相当于有机溶剂b的质量用量。
步骤(2)中所述的纯化的步骤优选如下:通过减压蒸馏除去溶剂,将所得粘稠液体用三氯甲烷溶解,用蒸馏水洗涤三至五次,然后减压蒸馏除去三氯甲烷,得到纯化后的pdms-b-(pdms-g-acys)。
步骤(3)中所述的pdms-b-(pdms-g-acys)与盐酸配比优选为按照pdms-b-(pdms-g-acys)中乙酰基的摩尔量:盐酸中氯化氢的摩尔量=1:4配比。
步骤(3)中所述的回流加热反应的条件优选为在75~90℃搅拌反应5h;更优选为于90℃搅拌反应5h。
步骤(3)中所述的有机溶剂d优选为甲醇、乙醇和异丙醇中的一种或至少两种。
步骤(3)中所述的有机溶剂d的质量用量优选为相当于盐酸质量的2.4~2.5倍。
步骤(3)中所述的纯化的步骤优选如下:减压蒸馏除去溶剂和氯化氢,将得到的粗产品溶解于水中,在蒸馏水中透析除去小分子杂质,得到纯化后的pdms-b-(pdms-g-cys)。
步骤(3)中所述的透析的条件优选为使用截留分子量为2000da的透析袋在蒸馏水中透析3天。
通过控制步骤(1)中d3、d3v单体的投料比、聚合反应时间,可以制备得到pdms嵌段与pvms嵌段长度可调,pvms嵌段中乙烯基链节比例可调的pdms-b-pvms,继而由后续反应得到不同嵌段长度比与不同半胱氨酸接枝比的pdms-b-(pdms-g-cys)。
一种接枝半胱氨酸的聚硅氧烷嵌段共聚物,通过上述制备方法得到。
所述的接枝半胱氨酸的聚硅氧烷嵌段共聚物优选为具有如下特征的接枝半胱氨酸的聚硅氧烷嵌段共聚物:嵌段长度比例为pdms:pdms-g-cys=2~4:8,pdms嵌段长1~2kda,pdms-g-cys嵌段长4kda。
所述的接枝半胱氨酸的聚硅氧烷嵌段共聚物在水溶液中制备固体材料表面抗蛋白吸附层中的应用。
所述的应用包括如下步骤:将固体材料置于上述接枝半胱氨酸的聚硅氧烷嵌段共聚物水溶液中浸泡,即可得到表面具有抗蛋白吸附层的固体材料。
所述的固体材料包括金属材料、高分子材料。
所述的浸泡的时间优选为30min。
本发明相对于现有技术具有如下的优点及效果:
本发明提供的接枝半胱氨酸的聚硅氧烷嵌段共聚物是含有两性离子的两亲性嵌段共聚物,由于在两性离子聚合物中引入疏水性作用结构,有利于两性离子聚合物在水溶液中向固-液界面吸附形成稳定的两性离子吸附层,发挥抗蛋白吸附作用。
附图说明
图1为实施例1的化学反应路线图。
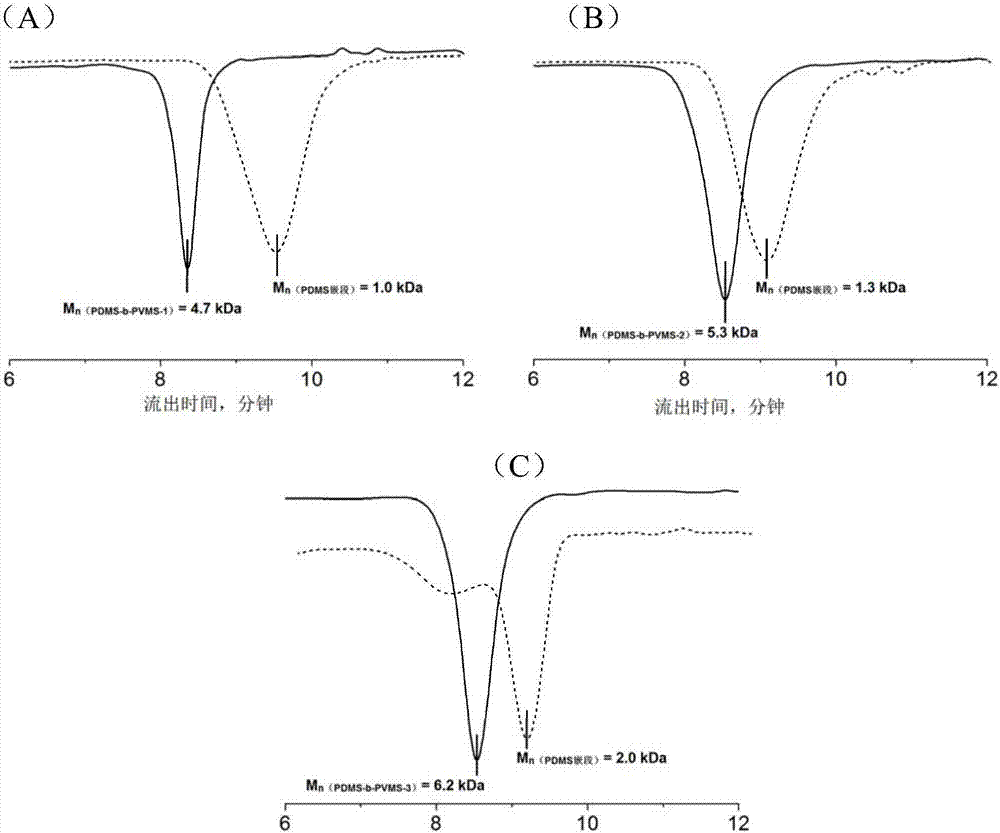
图2为pdms-b-pvms的gpc流出曲线图;其中,图(a)为实施例1制备的pdms-b-pvms-1,图(b)为实施例2制备的pdms-b-pvms-2,图(c)为实施例3制备的pdms-b-pvms-3。
图3为pdms-b-pvms的核磁共振氢谱图;其中,图(a)为实施例1制备的pdms-b-pvms-1,图(b)为实施例2制备的pdms-b-pvms-2,图(c)为实施例3制备的pdms-b-pvms-3。
图4为实施例1的pdms-b-(pdms-g-cys)-1的红外表征谱图。
图5为实施例1的pdms-b-(pdms-g-cys)的核磁共振氢谱与相应质子的化学位移示意图。
图6为在金片表面吸附了不同的pdms-b-(pdms-g-cys)后的牛血清蛋白吸附量检测结果图。
具体实施方式
下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
本发明所用的试剂均可从市场购得。
实施例1
聚二甲基硅氧烷-聚硅氧烷接枝半胱氨酸嵌段共聚物的制备:嵌段长度比例为pdms:pdms-g-cys=1:4,pdms嵌段长1kda,pdms-g-cys嵌段长4kda。pdms-g-cys嵌段中,其接枝半胱氨酸基团的链节所占总链节数比例(接枝比)为1/2。制备过程如图1所示,具体步骤如下:
(1)在装有磁力搅拌子的反应瓶中,加入2.5gd3,然后以硅胶塞封口,将瓶内抽真空后通入高纯氮气,重复操作三次以保证瓶中充满氮气。以注射器注入10ml干燥四氢呋喃溶解d3得到澄清溶液后,在室温下注入含2.5mol/l正丁基锂的正己烷溶液1.0ml,引发阴离子开环聚合。在0~5℃下保持搅拌约12h后,取少量反应液以甲醇终止活性链端,用于gpc测试第一嵌段(pdms)分子量,其结果如图2(a)中的虚线所示。取样后,注入含5.3gd3v与4.7gd3的干燥四氢呋喃溶液20ml,在0~5℃下继续反应约12h形成含有乙烯基甲基硅氧烷链节的第二嵌段pvms。取0.4ml三甲基氯硅烷注入到反应瓶中,继续搅拌约30min以完全终止聚合。将反应液旋转蒸发除去溶剂和小分子单体后,有大量氯化锂副产物粉末析出,过滤得到无色透明的粘稠液体产物pdms-b-pvms-1;
pdms-b-pvms-1中两嵌段的分子量可以由gpc(凝胶渗透色谱)测试得到,gpc测试条件为:以氯仿为流动相,设置流速为1.0ml/min,设置示差检测器温度为35℃,色谱柱分离温度为40℃。以窄分散聚苯乙烯作为标准样测定标准曲线并计算样品分子量与分子量分布。其结果如图2(a)所示,其中虚线是第一嵌段(pdms)的流出曲线,实线是二嵌段分子链pdms-b-pvms-1的流出曲线。最终产物的半胱氨酸基团接枝比即为pvms嵌段中含乙烯基的链节占总链节数的比例,可由核磁共振氢谱中相关峰的积分面积计算得到,其结果如图3(a)所示。核磁共振氢谱测试条件为:以氘代氯仿溶解样品,在25℃下测试。
(2)在装有磁力搅拌子的透明石英烧瓶中,加入1.0gpdms-b-pvms-1、1.05gn-乙酰半胱氨酸、0.38g安息香二甲醚;加入5.0g四氢呋喃和5.0g甲醇充分溶解上述原料形成无色透明溶液。在360nm波长的紫外光照射下,保持搅拌反应30min。结束反应后,减压蒸出溶剂,得到粘稠液体粗产物,将其溶于三氯甲烷中,以蒸馏水洗涤三次。最后减压蒸馏除去三氯甲烷,得到粘稠的液体产物pdms-b-(pdms-g-acys)-1;
(3)在装有磁力搅拌子的玻璃烧瓶中,加入1.0gpdms-b-(pdms-g-acys)-1,用2.63g乙醇溶解,加入1.05g质量浓度为36%的盐酸,在90℃下回流5h。结束反应后,减压蒸出乙醇、水与氯化氢,将粗产物溶解于水中,以截留分子量为2000da的透析袋在蒸馏水中透析3天。最后取透析袋中液体减压蒸溜除去水分,得到最终产物pdms-b-(pdms-g-cys)-1。
最终产物的红外表征结果如图4所示:其中,3415cm-1是-nh3+的伸缩振动吸收峰,1733cm-1是-coo-的伸缩振动吸收峰,1000–1100cm-1是si-o-si线性聚硅氧烷的特征吸收峰,表明产物中硅氧烷链、半胱氨酸等结构的存在。
最终产物的核磁表征结果如图5所示:产物用重水溶解,进行核磁共振氢谱测试,结果及相应化学位移的示意图如图所示。
实施例2
聚二甲基硅氧烷-聚硅氧烷接枝半胱氨酸嵌段共聚物的制备:嵌段长度比例为pdms:pdms-g-cys=3:8,pdms嵌段长1.5kda,pdms-g-cys嵌段长4kda。pdms-g-cys嵌段中,其接枝半胱氨酸基团的链节所占总链节数比例(接枝比)为1/2。
(1)在装有磁力搅拌子的反应瓶中,加入3.75gd3,然后以硅胶塞封口,将瓶内抽真空后通入高纯氮气,重复操作三次以保证瓶中充满氮气。以注射器注入10ml干燥四氢呋喃溶解d3得到澄清溶液后,在室温下注入含2.5mol/l的正丁基锂的正己烷溶液1.0ml,引发阴离子开环聚合。在0~5℃下保持搅拌约12h后,取少量反应液以甲醇终止活性链端,用于gpc测试第一嵌段(pdms)分子量,其结果如图2(b)的虚线所示。取样后,注入含5.3gd3v与4.7gd3的干燥四氢呋喃溶液20ml,在0~5℃下继续反应约12h形成含有乙烯基甲基硅氧烷链节的第二嵌段pvms。取0.4ml三甲基氯硅烷注入到反应瓶中,继续搅拌约30min以完全终止聚合。将反应液旋转蒸发除去溶剂和小分子单体后,有大量氯化锂副产物粉末析出,过滤得到无色透明的粘稠液体产物pdms-b-pvms-2;
pdms-b-pvms-2中两嵌段的分子量可以由gpc测试得到,gpc测试条件与实施例1一致。其结果如图2(b)所示,其中虚线是第一嵌段(pdms)的流出曲线,实线是二嵌段分子链pdms-b-pvms-2的流出曲线。最终产物的半胱氨酸基团接枝比即为pvms嵌段中含乙烯基的链节占总链节数的比例,可由核磁共振氢谱中相关峰的积分面积计算得到,核磁共振氢谱测试条件与实施例1一致,其结果如图3(b)所示。
(2)在装有磁力搅拌子的透明石英烧瓶中,加入1.1gpdms-b-pvms-2、1.05gn-乙酰半胱氨酸、0.38g安息香二甲醚;加入6.0g四氢呋喃和6.0g甲醇充分溶解上述原料形成无色透明溶液。在360nm波长的紫外光照射下,保持搅拌反应30min。结束反应后,减压蒸出溶剂,得到粘稠液体粗产物,将其溶于三氯甲烷中,以蒸馏水洗涤三次。最后减压蒸馏除去三氯甲烷,得到粘稠的液体产物pdms-b-(pdms-g-acys)-2;
(3)在装有磁力搅拌子的玻璃烧瓶中,加入1.0gpdms-b-(pdms-g-acys)-2,用2.53g乙醇溶解,加入1.02g质量浓度为36%的盐酸,在90℃下回流5h。结束反应后,减压蒸出乙醇、水与氯化氢,将粗产物溶解于水中,以截留分子量为2000da的透析袋在蒸馏水中透析3天。最后取透析袋中液体减压蒸溜除去水分,得到最终产物pdms-b-(pdms-g-cys)-2。
实施例3
聚二甲基硅氧烷-聚硅氧烷接枝半胱氨酸嵌段共聚物的制备:嵌段长度比例为pdms:pdms-g-cys=1:2,pdms嵌段长2kda,pdms-g-cys嵌段长4kda。pdms-g-cys嵌段中,其接枝半胱氨酸基团的链节所占总链节数比例(接枝比)为1/2。
(1)在装有磁力搅拌子的反应瓶中,加入5.0gd3,然后以硅胶塞封口,将瓶内抽真空后通入高纯氮气,重复操作三次以保证瓶中充满氮气。以注射器注入10ml干燥四氢呋喃溶解d3得到澄清溶液后,在室温下注入含2.5mol/l的正丁基锂的正己烷溶液1.0ml,引发阴离子开环聚合。在0~5℃下保持搅拌约12h后,取少量反应液以甲醇终止活性链端,用于gpc测试第一嵌段(pdms)分子量,其结果如图2(c)的虚线所示。取样后,注入含5.3gd3v与4.7gd3的干燥四氢呋喃溶液20ml,在0~5℃下继续反应约12h形成含有乙烯基甲基硅氧烷链节的第二嵌段pvms。取0.4ml三甲基氯硅烷注入到反应瓶中,继续搅拌约30min以完全终止聚合。将反应液旋转蒸发除去溶剂和小分子单体后,有大量氯化锂副产物粉末析出,过滤得到无色透明的粘稠液体产物pdms-b-pvms-3;
pdms-b-pvms-3中两嵌段的分子量可以由gpc测试得到,gpc测试条件与实施例1一致。其结果如图2(c)所示,其中虚线是第一嵌段(pdms)的流出曲线,实线是二嵌段分子链pdms-b-pvms-3的流出曲线。最终产物的半胱氨酸基团接枝比即为pvms嵌段中含乙烯基的链节占总链节数的比例,可由核磁共振氢谱中相关峰的积分面积计算得到,核磁共振氢谱测试条件与实施例1一致,其结果如图3(c)所示。
(2)在装有磁力搅拌子的透明石英烧瓶中,加入1.2gpdms-b-pvms-3、1.05gn-乙酰半胱氨酸、0.38g安息香二甲醚;加入6.0g四氢呋喃和6.0g甲醇充分溶解上述原料形成无色透明溶液。在360nm波长的紫外光照射下,保持搅拌反应30min。结束反应后,减压蒸出溶剂,得到粘稠液体粗产物,将其溶于三氯甲烷中,以蒸馏水洗涤三次。最后减压蒸馏除去三氯甲烷,得到粘稠的液体产物pdms-b-(pdms-g-acys)-3;
(3)在装有磁力搅拌子的玻璃烧瓶中,加入1.0gpdms-b-(pdms-g-acys)-3,用2.43g乙醇溶解,加入1.00g质量浓度为36%的盐酸,在90℃下回流5h。结束反应后,减压蒸出乙醇、水与氯化氢,将粗产物溶解于水中,以截留分子量为2000da的透析袋在蒸馏水中透析3天。最后取透析袋中液体减压蒸溜除去水分,得到最终产物pdms-b-(pdms-g-cys)-3。
应用实施例
抗蛋白吸附性能监测:设置蛋白质模型为牛血清蛋白;对照组为空白金片;实验组的金片在分别含3.0mg/ml实施例1-3所制备得到的pdms-b-(pdms-g-cys)-1、pdms-b-(pdms-g-cys)-2、pdms-b-(pdms-g-cys)-3的纯水溶液中浸泡30min以在表面形成对应嵌段共聚物的吸附层,然后依次用纯水、pbs缓冲液冲洗表面;对照组和实验组的金片大小相同。将空白金片与实验组金片分别称重后,再分别浸泡在相同体积的浓度为含1.0mg/ml的牛血清蛋白/pbs缓冲液溶液中30min,使蛋白吸附。吸附后以pbs缓冲液冲洗表面。以石英晶体微量天平来检测金片表面的蛋白吸附量。结果如图5所示。可见实施例1~3制备得到的聚二甲基硅氧烷-聚硅氧烷接枝半胱氨酸嵌段共聚物可以在水溶液中吸附到固体材料表面,形成稳定的吸附层,并显著的降低蛋白吸附量。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、
替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!