一种有机硼烷催化ε‑己内酯开环聚合的方法与流程
本发明属于高分子合成化学领域,具体涉及一种有机硼烷催化ε-己内酯开环聚合的方法。
背景技术:
脂肪族聚酯,如聚己内酯、聚乳酸和聚羟基脂肪酸酯,是一类非常具有应用前景的高分子材料。它们的聚合物链由一系列的酯键组成,在微生物、水或者霉的作用下,脂肪族聚酯可以发生降解反应,完全分解为小分子化合物。脂肪族聚酯不仅具有良好的生物可降解性和生物相容性,还具有优异的物理机械和加工性能,在生物组织工程材料、手术缝合线、医用螺钉和药用缓释材料等方面都有着重要的应用。
ε-己内酯的开环聚合是生产聚己内酯最重要的方法。目前,开环聚合的催化剂主要是锡、铝、稀土金属和过渡金属的配合物。然而,这些金属配合物成本较高且制备过程复杂,如专利cn105294745a。另外,金属催化剂具有一定的细胞毒性,而且在聚合物的分离纯化过程中无法将它们除去,这势必会限制脂肪族聚酯在生物领域的进一步应用。因此,有机非金属催化ε-己内酯的开环聚合成为业界关注的焦点。
许多课题组已经开发了各种类型的有机非金属催化剂,如二甲氨基吡啶(dmap)、n-杂环卡宾类(nhcs)、膦腈碱(t-bup4)等有机碱和甲基磺酸(ch3so3h)、三氟甲基磺酸(tfoh)等质子酸(参见macromolecules2010,43:2093)。虽然这些催化剂容易在聚合的后处理过程中除去,但是利用它们合成高分子量的线性聚己内酯时都必须加入引发剂(一般为醇类)。一方面加引发剂过程繁琐,另一方面聚己内酯上的引发剂端基也会对其性能产生影响。另外,现有的许多有机非金属催化剂不易保存,而且催化聚合时要求严格无水无氧的条件,如cn102093541a。因此,探索催化活性高、容易保存且聚合过程容易操作的有机非金属催化剂,对于ε-己内酯的开环聚合具有重要意义。三(五氟苯基)硼烷是一种非传统的路易斯酸,具有对水、氧稳定,使用方便的优点(参加化学通报,2007,1:34)。实验发现在60~120℃时三(五氟苯基)硼烷可以催化ε-己内酯的开环聚合,得到分子量分布较窄的聚己内酯,并且聚合过程容易操作。
技术实现要素:
在所论述的现有技术的背景下,本发明提供了一种有机硼烷催化ε-己内酯开环聚合的方法。该方法无需外加引发剂,具有反应较快、聚合过程简单的特点,而且聚合产物分子量较高、分子量分布较窄。
具体而言,本发明所用的有机硼烷为三(五氟苯基)硼烷,以ε-己内酯作为聚合单体,在不外加引发剂时发生开环聚合得到高分子量的聚酯。
本发明通过以下技术方案实现。
一种有机硼烷催化ε-己内酯开环聚合的方法,包括下列步骤:
(1)在惰性气体保护下或者在空气中,向反应瓶中依次加入三(五氟苯基)硼烷、反应溶剂和ε-己内酯,密封后反应;
(2)反应结束后,先用有机溶剂稀释反应液,再用冷却的无水甲醇沉淀,经过抽滤、真空干燥后得到聚酯。
优选的,步骤(1)所述惰性气体为氮气或者氩气。
优选的,步骤(1)所述反应溶剂为甲苯。
优选的,当进行本体聚合时,不需要加入反应溶剂。
优选的,步骤(1)所述ε-己内酯与三(五氟苯基)硼烷的摩尔比为(50~400):1。
优选的,步骤(1)所述反应的温度为60~120℃。
优选的,步骤(1)所述反应的时间为12~24小时。
优选的,步骤(2)所述有机溶剂为二氯甲烷、氯仿、甲苯或者四氢呋喃。
所述的三(五氟苯基)硼烷的结构通式如下:
所述的ε-己内酯的结构通式如下:
开环聚合为本体聚合或者在少量溶剂中进行溶液聚合,优先选用本体聚合。
当本方法选用溶液聚合时,所用的溶剂为甲苯。
本方法的反应温度可以为60~120℃,优选ε-己内酯与三(五氟苯基)硼烷为400:1下的本体聚合,反应时间为12小时。
本方法可以在惰性气体保护下或者在空气中进行,优先选用惰性气体保护。
本方法选用核磁共振氢谱(1hnmr)和基质辅助激光解析电离-飞行时间质谱(maldi-tofms)表征聚合产物的结构,选用凝胶渗透色谱(gpc)测定聚合产物的分子量和分子量分布。
提供下述试验部分以说明测试方法,但是在任何情况下不应将其理解为对本发明保护范围的限制。试验部分如下:
1hnmr测试时仪器型号为brukerav400,选用的溶剂为氘代氯仿(cdcl3),并且参照cdcl3的溶剂峰(7.25ppm)进行化学位移校正。
maldi-tofms测试时仪器型号为brukerautoflexiiismartbeam,选用的基质为2,5-二羟基苯甲酸(dhb);离子化试剂为碘化钠(nai);溶剂为四氢呋喃(thf),并且用标准混合多肽进行分子量校正。
gpc测试时仪器型号为waters2414,选用的溶剂为thf,并且用聚苯乙烯标样建立的标准曲线进行分子量和分子量分布校正。
与现有技术相比,本发明具有以下的有益效果:
(1)本发明公开了有机硼烷催化ε-己内酯开环聚合的方法,为聚己内酯的合成提供了新的选择。
(2)本发明选用三(五氟苯基)硼烷作为开环聚合的催化剂,制备的聚己内酯中不存在任何金属残留,突破了其在医用材料方面应用的限制。
(3)利用本方法进行ε-己内酯的开环聚合时,可以在惰性气体保护下或者空气中进行,并且无需外加引发剂,聚合过程简单,与现有的有机非金属催化体系相比,具有较大的优越性。
(4)本方法中三(五氟苯基)硼烷对ε-己内酯的开环聚合表现出较高的催化活性,并且收率较高,聚合产物也具有较窄的分子量分布。
附图说明
图1为有机硼烷催化ε-己内酯开环聚合得到的聚己内酯的1hnmr图。
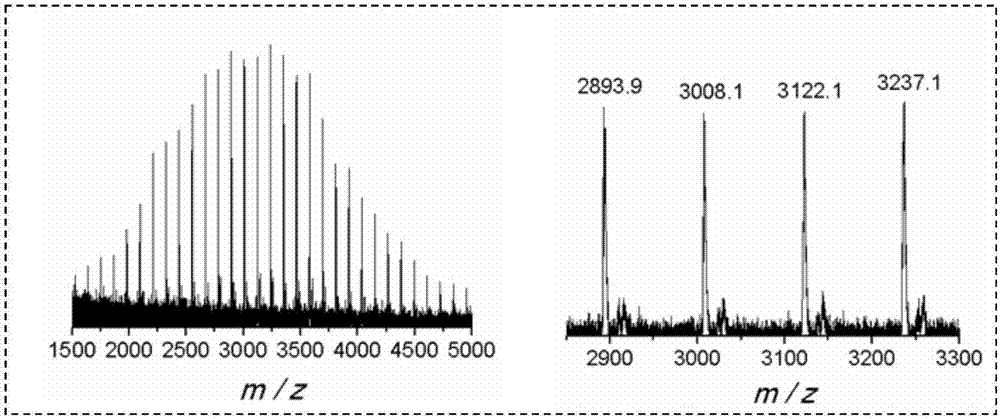
图2为有机硼烷催化ε-己内酯开环聚合得到的聚己内酯的maldi-tofms图。
图3为有机硼烷催化ε-己内酯开环聚合的化学反应路线图。
具体实施方式
下面结合实例与附图,用具体的实施例对本发明作进一步的说明,但本发明的保护范围不限于此。
本发明的有机硼烷催化ε-己内酯开环聚合的化学反应路线图如图3所示。
实施例1
向100ml的反应瓶中依次加入三(五氟苯基)硼烷(52mg,0.1mmol)、ε-己内酯(4.43ml,0.04mol),然后放入120℃的油浴锅中反应12小时。反应结束后,先加入25ml二氯甲烷稀释反应液,再用在-18℃下冷却的无水甲醇(300ml)沉淀。经过抽滤、真空干燥后得到聚合物3.1g,计算反应收率为68%。聚合物的mn为19800g/mol,pdi为1.72。
实施例2
在氮气的保护下,向100ml的反应瓶中依次加入三(五氟苯基)硼烷(52mg,0.1mmol)、甲苯(1ml)和ε-己内酯(4.43ml,0.04mol),然后放入120℃的油浴锅中反应12小时。反应结束后,先加入25ml二氯甲烷稀释反应液,再用在-18℃下冷却的无水甲醇(300ml)沉淀。经过抽滤、真空干燥后得到聚合物3.47g,计算反应收率为76%。聚合物的mn为22500g/mol,pdi为1.54。
实施例3
在氮气的保护下,向100ml的反应瓶中依次加入三(五氟苯基)硼烷(52mg,0.1mmol)、ε-己内酯(0.55ml,0.005mol),然后放入60℃的油浴锅中反应24小时。反应结束后,先加入3ml二氯甲烷稀释反应液,再用在-18℃下冷却的无水甲醇(50ml)沉淀。经过抽滤、真空干燥后得到聚合物0.54g,计算反应收率为95%。。聚合物的mn为4800g/mol,pdi为1.21。
采用1hnmr和maldi-tofms表征了本实施例所得聚合产物的结构,分别如图1和图2所示。从图1可以看出,在化学位移为3.64ppm处出现了聚己内酯末端羟基质子的特征峰。结合图2与表1可以看出,maldi-tofms测得的荷质比(m/z)大致等于ε-己内酯重复单元的质量数(dp×114.14)与钠离子的质量数(23)之和加上18。水的质量数刚好为18,因此可以推断聚合时的真正引发剂为体系中的微量水分。三(五氟苯基)硼烷在沉淀过程中容易除去,并且聚合产物中不存在常用的醇类引发剂的端基,本方法具有较大的优越性。其它实施例所得聚合产物的1hnmr和maldi-tofms图参见图1、图2。
表1
实施例4
在氮气的保护下,向100ml的反应瓶中依次加入三(五氟苯基)硼烷(52mg,0.1mmol)、ε-己内酯(1.11ml,0.01mol),然后放入80℃的油浴锅中反应20小时。反应结束后,先加入7ml二氯甲烷稀释反应液,再用在-18℃下冷却的无水甲醇(100ml)沉淀。经过抽滤、真空干燥后得到聚合物1.06g,计算反应收率为93%。。聚合物的mn为9800g/mol,pdi为1.28。
实施例5
在氮气的保护下,向100ml的反应瓶中依次加入三(五氟苯基)硼烷(52mg,0.1mmol)、ε-己内酯(2.22ml,0.02mol),然后放入80℃的油浴锅中反应20小时。反应结束后,先加入13ml二氯甲烷稀释反应液,再用在-18℃下冷却的无水甲醇(200ml)沉淀。经过抽滤、真空干燥后得到聚合物2.05g,计算反应收率为90%。。聚合物的mn为16300g/mol,pdi为1.37。
实施例6
在氮气的保护下,向100ml的反应瓶中依次加入三(五氟苯基)硼烷(52mg,0.1mmol)、ε-己内酯(4.43ml,0.04mol),然后放入120℃的油浴锅中反应12小时。反应结束后,先加入25ml二氯甲烷稀释反应液,再用在-18℃下冷却的无水甲醇(300ml)沉淀。经过抽滤、真空干燥后得到聚合物3.83g,计算反应收率为84%。。聚合物的mn为31700g/mol,pdi为1.42。
- 还没有人留言评论。精彩留言会获得点赞!