一种棉花GbDRP66319基因、编码蛋白及应用的制作方法
本发明属于生物工程领域,具体地说,涉及一种棉花gbdrp66319基因、编码蛋白及应用。
背景技术:
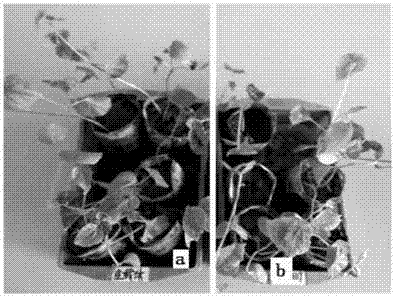
:棉花黄萎病是棉花生产中的主要病害之一,对棉花生产造成严重的影响和损失,病原体从植物的根部或根茎中入侵,并寄生在维管束中。被感染的棉株叶片发黄、枯萎、脱落,根和茎的维管束也均变褐色,棉铃变小,脱铃率高,甚至导致整个植株枯死,严重影响棉花的产量和品质。1891年,棉花黄萎病首次在美国被发现。随后,此病害迅速传播到全世界的主要棉花产区。1935年,我国通过引入美国棉种而引入该病原菌,然后随着棉花种子的运输而继续传播。目前,该病已经扩展到中国18个省、自治区的478个县(市)。许多棉花产区均出现该病害,并且中国北方产区比南方产区发病情况严重。该病导致棉花产量损失占多年平均产量的15-20%,在发病严重年份损失量可达50%,甚至造成绝产。上世纪90年代,该病的蔓延速度加剧。据调查估测,1995和1996年,每年大约2667000hm2的棉田遭受病害,皮棉产量损失约100000吨。2002年发病棉田已超过30000000hm2。因此,棉花黄萎病已成为影响我国棉花高产稳产的主要障碍。目前该病害的控制管理方法主要以防为主,主要有化学防治、利用农业措施、生物防治、选用抗病品种等。经过长期实践表明,利用化学、生物等方法防治棉花黄萎病均存在一定的局限性,选育和种植抗病品种才是最直接、最经济、最有效的措施。但是经过多年抗性鉴定结果表明,国内现存的棉花种质资源中对黄萎病高抗的材料较少,并且大都为海岛棉,陆地棉材料中70%以上为感病材料,达到高抗的材料不足1%。。因此,高抗材料的缺乏是限制抗棉花黄萎病育种的主要因素。利用常规育种方法对棉花抗病性进行遗传改良,不仅育种周期长,而且效率低。技术实现要素:有鉴于此,本发明针对上述的问题,提供了一种棉花gbdrp66319基因、编码蛋白及应用。为了解决上述技术问题,本发明公开了一种棉花gbdrp66319基因的编码蛋白,其具有seqidno.2所示的氨基酸序列或该序列经替换、缺失或添加一个或几个氨基酸形成的具有同等功能的氨基酸序列。本发明还提供一种编码上述蛋白的基因。进一步地,其具有seqidno.1所示的核苷酸序列。本发明还公开了一种含有上述基因的载体。本发明还公开了一种含有上述载体的宿主细胞。本发明还公开了一种含有上述基因的转化植物细胞。本发明还公开了一种上述基因在控制植物抗黄萎病中的应用。进一步地,所述的植物为棉花。进一步地,所述的植物为拟南芥。与现有技术相比,本发明可以获得包括以下技术效果:1)本发明所获得的gbdrp66319基因不仅与抗黄萎病qtl位点(nau3592)紧密连锁,同时该基因在黄萎病菌诱导后,在mrna转录水平上表现出基因表达的变化,说明本发明的gbhir66319基因与黄萎病抗性高度相关。2)gbdrp66319在ncbi中blast搜索,未发现在其他物种中存在与之同源的基因,推测该基因可能是棉属中所特有的一类基因,并且unigene66319基因包含p-loop,kinase2,kinase3,glpl、mhdl五个motif,以及lrr_bac,lrr_cc和lrr_typ等抗病基因的保守结构域,说明该基因是一个典型的抗病基因(r基因),可以产生抗病蛋白。表达分析表明unigene66319在黄萎病菌诱导4h、12h、24h、48h后持续上调表达,暗示gbdrp66319基因表达量高的棉花植株具有更好的抗病机制,gbdrp66319基因表达量高的棉花植株会产生更多的抗病蛋白,从而快速激发抗病反应。3)gbdrp66319基因沉默表达后对黄萎病菌抗性减弱,并且gbdrp66319基因沉默表达后感病陆地棉比抗病海岛棉的发病情况较重,说明gbdrp66319基因的表达有助于棉花对黄萎病的抗性,因此,转gbdrp66319基因的转基因棉花对黄萎病的抗性增强。当然,实施本发明的任一产品并不一定需要同时达到以上所述的所有技术效果。附图说明此处所说明的附图用来提供对本发明的进一步理解,构成本发明的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:图1是本发明棉花根部组织总rna电泳图,图中,m为分子量marker,1自上到下分别为rna的28s、18s、5s带型;图2是本发明利用unigene66319两端的5’非编码区和3’非编码区的特异性引物对海岛棉cdna进行基因扩增图,其中,1为扩增出的目的片段,即unigene66319编码基因片段,m为分子量marker;图3是本发明unigene66319目的基因片段与克隆t载体连接后,大肠杆菌在含有卡那霉素的lb培养基中培养过夜后长出的阳性白色克隆产物;图4是本发明unigene66319在黄萎病菌诱导12h后在海岛棉和陆地棉中的组织表达特异性荧光定量pcr分析;图5是本发明gbdrp66319蛋白质的磷酸化位点预测;图6是本发明gbdrp66319蛋白质的跨膜结构域分析;图7是本发明gbdrp66319及其同源基因的保守结构域分析;图8是本发明gbdrp66319基因的系统发育树分析;图9是本发明pbi121-unigene66319植物重组表达载体在含有卡那的lb培养基中过夜培养后长出白色阳性克隆产物;图10是本发明pbi121-unigene66319植物重组表达载体的pcr检测,其中,m为分子量marker,1-3均为pcr产物;图11是本发明含有unigene66319的重组质粒为模板,扩增获得目的基因vigs片段,其中,m为分子量marker,1-2均为目的基因vigs片段;图12是本发明重组质粒和病毒载体trv双酶切图,其中,图a中m为分子量marker,1-2均为重组质粒双酶切产物。图b中m为分子量marker,1为病毒载体trv双酶切产物;图13是本发明unigene66319目的基因vigs片段与病毒载体trv连接后在含有卡那霉素的lb培养基中长出白色克隆产物;图14是本发明注入含pyl156-pds载体vigs悬浮液的棉花阳性对照植株在注射一周后真叶变化情况;图15是本发明含pyl156空载体、pyl156-66319的vigs海岛棉3-79植株;图a为注入含pyl156空载体vigs悬浮液的棉花植株,图b为注入含pyl156-66319载体vigs悬浮液的棉花植株;图16分别为含pyl156空载体、pyl156-66319的vigs陆地棉tm-1植株;图a为注入含pyl156空载体vigs悬浮液的棉花植株,图b为注入含pyl156-66319载体vigs悬浮液的棉花植株。具体实施方式以下将配合实施例来详细说明本发明的实施方式,藉此对本发明如何应用技术手段来解决技术问题并达成技术功效的实现过程能充分理解并据以实施。实施例11、海岛棉总rna的提取及cdna的合成本发明采用改良的异硫氰酸胍法提取棉花子叶、下胚轴及根部的总rna,具体步骤如下:1)rna的提取:1.1)在干净的50ml离心管中加入15ml已配好的rna提取液及150μl的β-巯基乙醇,混匀后放在冰上。1.2)先在灭菌后的研钵内加入液氮,然后加入1.5g左右的样品,迅速研磨成白色粉末状,用药匙刮下研钵上的样品,将磨碎的样品加入含有提取液的离心管中,混匀后加入1.5ml2mol·l-1的乙酸钠缓冲液(ph4.2),剧烈震荡混匀后放冰上。1.3)等体积加入氯仿,剧烈震荡混匀后置于冰上15min。1.4)将离心管放于低温离心机内,12000r·min-1,4℃离心15min,将上清液小心的转移到另一50ml离心管中,加入等体积的氯仿,剧烈摇匀后放置冰上15min。1.5)12000r·min-1,4℃离心15min,取上清液于心的离心管中,再加入等体积冰冷的异丙醇,-20℃沉淀30min以上。1.6)12000r·min-1,4℃离心15min,离心管底部出现白色沉淀。倒掉上清液,加入1/3体积的提取液,充分溶解后,加入等体积的氯仿,混匀不分层,放置冰上15min。1.7)12000r·min-1,4℃离心15min,取上清液并分装至7ml离心管中,再加入等体积的冰冷的异丙醇,-20℃沉淀30min以上。1.8)12000r·min-1,4℃离心15min,离心管底部出现白色沉淀,弃上清液,75%depc处理的乙醇进行洗涤后,转移至1.5ml离心管中,风干。1.9)加入400μldepc水溶解rna。2)rna的纯化:2.1)加入等体积的酚:氯仿(1:1)400μl,涡旋、混匀,13000r·min-1,4℃离心3min。2.2)取上清液,加入等体积的氯仿,涡旋、混匀,冰上静置10min,13000r·min-1,4℃离心10min。2.3)取上清液,加入1/10体积2mol·l-1的乙酸钠溶液和2.5体积无水乙醇,-20℃沉淀30min以上。13000r·min-1,4℃离心10min,2.4)弃上清,用75%depc处理的乙醇洗涤rna沉淀2次,风干。2.5)加入30~40μl的depc水溶解rna沉淀,-70℃保存。以海岛棉3-79根部组织为材料进行总rna提取,经1.0%琼脂糖凝胶电泳显示如图1,rna的28s、18s、5s带型清晰,无降解。然后使用nanodrop2000微量紫外分光光度计检测rna的质量,a260/a280=1.92,说明无蛋白质污染。a260/a230=2.08,无其他杂离子残留。因此,提取的rna的质量和纯度符合实验要求,能够进行cdna反转录。2、cdna的合成cdna的合成采用反转录试剂盒primescripttmrtreagentkitwithgdnaeraser(takara公司)。操作方法如下:步骤1:去除基因组dna反应试剂使用量5×gdnaeraserbuffer2.0μlgdnaeraser1.0μltotalrna*1rnasefreedh2oupto10μl反应条件:42℃2min(或者室温5min*2),4℃保存。步骤2反转录反应试剂使用量步骤1的反应液10.0μlprimescriptrtenzymemixi1.0μlrtprimermix1.0μl5×primescriptbuffer24.0μlrnasefreedh2o4.0μltotal20μl反应条件:37℃15min,85℃5sec,4℃保存。3、基因片段的分离与获得为了克隆unigene66319完整的编码序列(codingsequence,cds),将转录组数据库中unigenes66319序列与雷蒙德氏棉的cds数据库进行本地比对,获得与之序列相似性最大的序列,然后以其两端的5’非编码区和3’非编码区设计特异性引物,利用primer5.0设计引物,由立菲公司合成。引物如表1所示:表1unigene66319的特异性引物以海岛棉3-79的cdna为模板,使用新合成的特异引物进行基因扩增,合成目的片段,即unigene66319完整的编码序列cds。pcr反应体系为50μl:模板cdna5μl,primer(10μmol·l-1)各1μl,2xtranstaqpcrsupermix25μl,ddh2o18μl。pcr反应程序为:94℃预变性10min;94℃变性30s,50℃30s,72℃90s,反应35个循环;72℃延伸10min,4℃保存。4、目的片段胶回收若上述步骤获得的pcr产物无非特异性扩增,进行dna纯化后可直接与克隆t载体连接。若pcr扩增获得的目的片段有非特异性片段,则需先进行胶回收,去除杂带后再与t载体连接。胶回收具体步骤如下:1)使用tae缓冲液制作1.0%的琼脂糖凝胶,然后对目的dna进行琼脂糖凝胶电泳。2)在紫外灯下切出含有目的dna的琼脂糖凝胶,用纸巾吸尽凝胶表面的液体。此时应注意尽量切除不含目的dna部分的凝胶,尽量减小凝胶体积,提高dna回收率。胶块超过300mg时,请使用多个column进行回收,否则严重影响收率。注:切胶时请注意不要将dna长时间暴露于紫外灯下,以防止dna损伤。3)切碎胶块。胶块切碎后可以加快操作步骤6)的胶块溶解时间,提高dna回收率。4)称量胶块重量,计算胶块体积。计算胶块体积时,以1mg=1μl进行计算。5)向胶块中加入胶块3倍体积的溶解液buffergm。6)均匀混合后室温15-25℃溶解胶块,此时应间断振荡混合,使胶块充分溶解(约5~10分钟)。注:胶块一定要充分溶解,否则将会严重影响dna的回收率。高浓度凝胶可以适当延长溶胶时间。7)当凝胶完全溶解后,观察溶胶液的颜色,如果溶胶液颜色由黄色变为橙色或粉色,向上述胶块溶解液中加入3m醋酸钠溶液(ph5.2)10μl,均匀混合至溶液恢复黄色。当分离小于400bp的dna片段时,应在此溶液中再加入终浓度为20%的异丙醇。8)将试剂盒中的spincolumn安置于collectiontube上。9)将上述操作步骤7)的溶液转移至spincolumn中,12,000rpm离心1分钟,弃滤液。注:如将滤液再加入spincolumn中离心一次,可以提高dna的回收率。10)将700μl的bufferwb加入spincolumn中,室温12,000rpm离心30秒钟,弃滤液。注:请确认bufferwb中已经加入了指定体积的100%乙醇。11)重复操作步骤10)。12)将spincolumn安置于collectiontube上,室温12,000rpm离心1分钟。13)将spincolumn安置于新的1.5ml的离心管上,在spincolumn膜的中央处加入30μl灭菌蒸馏水或elutionbuffer,室温静置1分钟。注:将灭菌蒸馏水或elutionbuffer加热至60℃使用时有利于提高洗脱效率。14)室温12,000rpm离心1分钟洗脱dna。15)胶回收后的目的片段进行琼脂糖凝胶电泳,检测胶回收效率。5、目的片段与克隆t载体连接克隆t载体选用的是-t5zerocloningvector,该载体通过自杀基因表达与否筛选阳性重组子,当目的片段与t载体连接成功时,阻止自杀基因正常表达,那么包含重组载体的大肠杆菌就可以正常生长,当目的片段与载体未连接成功时,自杀基因就会正常表达,包含该载体的大肠杆菌就无法正常生长。该载体包含卡那霉素和氨苄青霉素两种筛选标记。连接体系:试剂使用量目的片段(胶回收后)4μlpeasy-t5zerocloningvector1μl轻轻混合,室温(20℃-37℃)反应5分钟。反应结束后,将离心管置于冰上。注:1、最佳插入片段dna量载体与片段摩尔比=1:7,可以粗略的按照“1kb20ng”的比例计算。(如1kb加20ng、1.5kb加30ng等)2、最佳载体使用量:1μl3、最佳反应体系3-5μl,体积不足时可以补充无菌水连接反应时间:注:片段为胶回收产物时,反应时间取最大值。连接反应的温度:最佳连接温度为25℃,最好采用pcr仪控温(上盖温度设为40℃)。目的片段与克隆t载体连接成功后,阻止t载体上自杀基因的表达,大肠杆菌则可以正常生长繁殖,如图3为大肠杆菌在含有卡那霉素的lb培养基中培养过夜后长出的白色克隆。挑取上述白色克隆进行pcr检测其是否为阳性克隆,选取阳性克隆送去立菲公司测序,其核苷酸序列如seqidno.1所示,其氨基酸序列如seqidno.2所示。6、重组t载体向大肠杆菌dh5α感受态转化1)加连接产物于50μldh5α感受态细胞中(在感受态细胞刚刚解冻时加入连接产物),轻弹混匀,冰浴20-30分钟。2)42℃热激30秒,立即置于冰上2分钟。3)加250μl平衡至室温的lb液体培养基,200转、37℃孵育1小时。4)取200μl菌液铺板(lb固体培养基中加抗生素氨苄),培养过夜(为得到较多克隆,4000rpm离心1min,弃掉部分上清,保留100-150μl,轻弹悬浮菌体,取全部菌液涂板,37℃恒温培养箱中培养过夜)。注:1llb培养基需加10g蛋白胨,5g酵母粉,10gnacl;若配制固体培养基需再加入15g琼脂粉。7、阳性重组子的鉴定和测序1)用灭菌牙签挑取白色菌落于10μl无菌水中,涡旋混合。2)25μlpcr反应体系中取1μl混合菌液作模板,用通用引物m13f和m13r鉴定重组子。3)pcr反应条件:94℃预变性10分钟,94℃变性30秒,55℃退火30秒,72℃延伸(根据片段的大小确定延伸时间)30个循环,72℃后延伸10分钟。4)pcr产物用琼脂糖凝胶电泳进行检测。经琼脂糖凝胶电泳检测为阳性的克隆,加入1mllb液体培养基(加氨苄)中进行扩大培养,200转、37℃,培养6h后,取500μl至1.5ml离心管(用封口膜封好)送至公司测序,测序引物用通用引物m13f,m13r。3.1.3.7组织特异性分析1.研究黄萎病菌诱导12h后棉花根、茎、子叶中基因表达特异性,总rna的提取及cdna的合成方法同上。2)荧光定量引物如表2:表2unigenes66319荧光定量引物序列3)荧光定量分析以actin基因为内参基因,分别利用特异性引物进行荧光定量pcr分析两个品种根、下胚轴和子叶片组织的抗性相关基因在枯萎病病原菌诱导下的表达量。通过荧光定量pcr分析unigene66319的组织表达特异性,结果如下图4所示:unigene66319黄萎病菌诱导12h后的海岛棉3-9下胚轴中表达量最高,在陆地棉tm-1根部组织中表达量最高。unigene66319是与抗黄萎病qtl附近的r基因高度同源的一类基因,转录组中注释信息显示其为包含bedfinger-nbs-lrr的抗性蛋白。通过荧光定量表达分析结果表明,该基因在黄萎病诱导后4-48h均上调表达;这个基因很有可能在棉花抗黄萎病中发挥重要的作用,因此,本实验克隆其完整的基因编码区,为进一步验证该基因功能打下基础。通过荧光定量pcr分析这两个基因的组织特异性表达情况,结果表明,同一基因在不同品种中的根、胚轴、子叶中的表达量不同。实施例2gbdrp66319基因的生物信息学分析根据unigene66319基因的注释信息,本发明命名为gbdrp66319。1、ghdrp66319蛋白序列的理化特性分析利用computepi/mw软件和protparam软件(http://web.expasy.org/protparam/)预测ghdrp66319蛋白序列的等电点和分子量及相关理化性质,利用netphos2.0(http://www.cbs.dtu.dk/services/netphos/)预测磷酸化位点,采用tmhmm软件预测蛋白的跨膜螺旋区,采用targetp对蛋白进行亚细胞定位预测。利用protparam软件(http://web.expasy.org/protparam/)分析gbdrp66319蛋白的理化性质,结果表明leu、ser、val、lys、pro含量最高,分别达到13.4%、10.3%、7.2%、6.8%、6.8%。带正电荷的氨基酸数目与带负电的氨基酸数目相同,均为29个,因此,该蛋白质本身不带电。该蛋白质的不稳定系数为48.67,属于不稳定蛋白。亲疏水性平均系数(gravy)为-0.026。负值表示该蛋白亲水性较强。磷酸化分析结果表明,ser磷酸化位点有6个、thr磷酸化位点有5个、tyr磷酸化位点1个,如图5所示。通过tmhmm软件分析跨膜结构域,如图6所示,该蛋白没有跨膜结构域,不是跨膜蛋白。通过targetp对蛋白进行亚细胞定位,未发现其有信号肽。因此推测该蛋白可能存在细胞质内发挥作用。2、gbdrp66319同源序列及保守结构域分析首先利用本实验克隆得到的gbdrp66319的完整编码序列与雷蒙德氏棉d组和亚洲棉a组的cds数据库进行比对,获得与之序列相似性较高的同源r基因。利用pfamdatabase和smart蛋白序列分析软件,interproscan软件鉴定其结构域和motif。然后利用dnaman软件进行多序列联配。gbdrp66319的cds与雷蒙德氏棉和亚洲棉基因组cds数据库比对,获得与之同源的nbs-lrr类基因,如表3所示,其中d组中有23个,a组中有6个。根据smart和interproscan等蛋白结构域预测软件,分析蛋白的保守结构域及motif,并鉴定其基因类型,如表3所示,利用dnaman对其同源基因进行联配,结果如图7所示,可以看出大部分基因包含p-loop,kinase2,kinase3,glplandmhdl五个motif,以及lrr_bac,lrr_cc和lrr_typ等抗病基因的保守结构域。表3gbdrp66319基因比对二倍体棉花雷蒙德氏棉(gossypiumraimondii)和亚洲棉(gossypiumarboreum)基因组数据库3、系统发育树分析利用mega5.1软件中的clustalw进行多序列联配后,然后使用邻接法(neighbor-joining,nj)构建进化树,对棉花中与ghdrp66319同源的蛋白进行分组。对gbdrp66319基因及与之同源的a组和d组基因共30个基因构建系统发育树,如图8所示,根据聚枝关系,将棉花中此类基因大致划分为6组。其中,3组和4组中既包含a组基因,又有d组中的基因;1组、2组、5组和6组中只含有d组特有的同源基因。故在此推测:3组和4组中可能为祖先基因,1组、2组、5组和6组中的d基因组上的基因可能是由于在a组和d组分化以后,d基因组发生扩增事件。同时也可以看出,gbdrp66319基因与cotton_gene_10001688的亲缘关系最近,推测海岛棉中的gbdrp66319基因是由雷蒙德氏棉中的cotton_gene_10001688基因进化而来。通过对gbdrp66319蛋白的理化特性分析发现,gbdrp66319蛋白可能的磷酸化位点丝氨酸有6个,苏氨酸有5个,酪氨酸有1个。蛋白磷酸化是细胞内最常见的一种蛋白修饰方式,蛋白通过磷酸化和去磷酸化调节是细胞信号传递中的重要环节,可以调控基因的表达、细胞的生长和分化等。通过对蛋白的跨膜结构分析,这两个蛋白不存在跨膜结构域,它们均为亲水性蛋白,并且通过亚细胞定位,它们均未有信号肽,因此,它们并非分泌蛋白,并且也不存在线粒体和叶绿体中,推测它们应该是在细胞质内发挥作用的一类蛋白。实施例3gbdrp66319基因功能的初步验证1、重组表达载体的构建1)引物的设计本实验采用in-fusion技术构建重组植物表达载体,该技术是在克隆引物的两端分别引入与表达载体插入位点两端各15个碱基,然后通过同源融合,使目的基因正确插入植物表达载体中。分别根据gbdrp66319基因的非编码区设计特异引物,在特异性上下游引物5,端各加上15个碱基,15个碱基分别为bamhi和saci酶切位点两侧的15个碱基。在线设计引物网址为http://bioinfo.clontech.com/infusion/convertpcrprimersinit.do引物序列见下表4。表4用于扩增gbdrp66319基因编码区的引物序列2)gbdrp66319基因编码区的获得以海岛棉3-79的cdna为模板,以上步骤设计的引物进行pcr,扩增出uingene66319两个基因的cds编码区。pcr反应体系及反应程序同第三章基因克隆。pcr产物经过琼脂糖凝胶电泳,若没有杂带,则可以经过纯化后直接进行下一步实验;若有杂带,则需要先进行胶回收。步骤与同源序列克隆时胶回收过程。3)pbi121-uingene66319植物重组表达载体的构建首先用限制性内切酶bamhi和xhoi双酶切植物表达载体pbi121,将载体中gus基因酶切下来,酶切体系共50μl如下:sacⅰ1μl,bamhⅰ1μl,pbi12120μl,10xnebuffer5μl,灭菌水23μl。反应时间3h或过夜,反应温度37℃。然后将上步扩增出的目的片段同酶切后的表达载体进行连接,连接体系如下:5xin-fusionhdenzymepremix1μl,酶切后的121表达载体1μl,目的片段3μl。反应条件:50℃15min(推荐pcr仪控温,上盖55℃),然后置于冰上。转化大肠杆菌dh5α,经卡那霉素(kan)筛选后,挑取生长良好的克隆进行pcr检测。pcr检测时用的引物为35s和nosr,构建的植物表达载体命名为pbi121-66319。4)农杆菌lba4404感受态的制备4.1)挑取lba4404单菌落在1mllb培养基,28℃,200rpm,过夜培养。4.2)取2ml农杆菌加入50mllb培养基,28℃,200rpm,培养至od值为0.5-0.8。5000rpm4℃离心5min。4.3)用10ml冰冷的0.15mnacl悬浮细胞,5000rpm4℃离心5min。4.4)用1ml冰冷的20mmcacl2在冰上悬浮,加75ul7%的dmso(每1ml加75ul)4.5)每管100ul分装,-70℃保存。5)重组表达载体pbi121-uingene66319转化农杆菌转化步骤如下:5.1)取感受态细胞50ul,冰上解冻,然后加入2-3ug重组质粒(6-10ul),冰上30min。5.2)液氮冷却1min,37℃水浴4min,冰上2min。5.3)重复步骤2一次,加1mllb,28℃200rpm摇菌4-6h。5.4)离心后去掉上清,保留200ul,悬浮菌体,涂lb板(加卡那和利福平),铝箔纸包裹,28℃暗培养两天。5.5)挑取单克隆,用通用引物35s和nosr进行pcr检测。6)pbi121-unigene66319植物重组表达载体的构建目的片段与酶切好的pbi12载体进行融合连接,转入大肠杆菌dh5α后,在含有卡那的lb培养基中过夜培养后长出白色克隆,如图9所示。挑取单克隆进行阳性鉴定,结果如图10所示,片段大小均为预期大小。可以进行后续转化农杆菌实验。2、vigs载体pyl156-66319的构建本发明选择的病毒载体为烟草脆裂病毒trv,载体选用pyl156载体。1)引物设计:引物设计要求目的片段大小300-500bp,且选择的酶切位点必须是病毒载体上含有,但目的片段上不含有,本实验选择的酶切位点为酶切位点选择bamh1和xho1,上下游引物5,端加上酶切位点和保护碱基。引物为表5用于扩增目的基因vigs片段的引物序列2)扩增目的基因vigs片段以含有gbdrp66319的重组质粒模板,及合成的特异性引物pcr扩增出目的片段。pcr反应体系及反应程序同上述获得目的基因时的程序。pcr产物经过琼脂糖凝胶电泳,若没有杂带,则可以经过纯化后直接进行下一步实验;若有杂带,则需要先进行胶回收。步骤同上。3)pyl156-66319vigs载体的构建将上步目的片段首先连接到克隆载体t5上,构建中间重组质粒t5-66319,转入大肠杆菌dh5α,送至公司测序。然后提取大肠杆菌中的重组质粒,再用xhoⅰ和bamhⅰ对其双酶切,得到两端具有粘性末端的目的基因,并和同样酶切的trv载体用t4连接酶进行连接,转化大肠杆菌dh5α,经卡那霉素筛选后,挑取生长良好的克隆进行pcr检测和酶切鉴定。最后构建的植物表达载体命名为pyl156-66319。双酶切体系共50ul:10*kbuffer5ul,bamhⅰ1ul,xhoⅰ1ul,载体(质粒)20ul,灭菌ddh2o23ul。连接过程:将质粒载体dna与插入dna片段混合制备成体积为5-10μl的dna溶液。载体dna和插入dna的摩尔数比一般为:0.03pmol:0.03-0.3pmol。向上述dna溶液中加入等体积(5-10μl)的solutioni,充分混匀。16℃反应30分钟。3、pyl156-66319载体转化农杆菌gv31011)重组质粒的提取提取方法同质粒提取试剂盒操作说明。2)农杆菌gv3101感受态的制备制备感受态操作方法同同制农杆菌lba4404。3)重组质粒转化农杆菌的步骤。3.1)准备超净工作台、常温离心机、28度恒温摇床及培养基、37°水浴锅、灭菌枪头、无菌玻璃珠、黑色塑料布、液氮、冰块。3.2)自-80度冰箱取出感受态,冰浴缓慢融化约10min待待转化质粒同样于冰浴中冷却。3.3)用预冷的枪头将2-5ug质粒加入刚融化的感受态中,轻轻弹碰混匀后水浴30min;3.4)液氮冷冻处理1-3min,37度快速热激3-5min,迅速与冰浴中放置约2min,重复该步骤;3.5)加1000微升空白lb培养液,封口膜封口,28度,180rpm,活化4-6h;3.6)5000rpm离心2min富集菌液,弃去上层多余培养基,管内余约200微升,无菌枪头缓慢吹打至沉沉菌重现悬浮;3.7)在超净台内风干三抗板,用无菌的玻璃珠或涂抹棒将菌液均匀涂布与平板上,风干,封口;3.8)用黑色塑料布遮光,倒置,28度培养24h左右;3.9)挑取单菌落用于扩摇、检测、保存等后续实验;以含有unigene66319cds的重组质粒为模板,扩增获得目的片段,经过检测,如图11和12所示,可知其片段大小符合要求,胶回收后可以进行下一步的连接转化实验。先将vigs片段与t5载体构建中间重组载体,转入大肠杆菌dh5α后,将阳性克隆送去公司测序检测,将含有目的片段的重组质粒提取出来,用xhoⅰ和bamhⅰ分别双酶切重组质粒和病毒载体trv,然后将二者用t4连接酶连接起来,转入大肠杆菌dh5α,在含有卡那霉素的lb中过夜培养,长出白色克隆如图13所示。4、vigs棉花植株的获得本发明采用pyl156-pds载体使植物中的叶绿体基因沉默表达,出现白化叶片,作为阳性对照。pyl156空载体作为阴性对照。1)包含pyl156-66319、pyl156-pds、pyl156-192及pyl156空载体的农杆菌分别在含有卡那霉素(50ug/ml)、庆大霉素(50ug/ml)、利福平(25ug/ml)的lb培养基上培养,活化菌体后,培养至od=1.5。2)4000rp离心5min。3)用vigs悬浮液将菌液调至od=1.5。25℃静置4h后,将pyl156-66319、pyl156-pds、pyl156空载体vigs悬浮液分别于pyl156-192的vigs悬浮液1:1混合。4)用无菌注射器对生长10天左右的棉花子叶注射,将vigs液体打满棉花子叶的下面。5)将注射后的棉花幼苗在25℃光照培养箱中培养。5、gbdrp66319基因功能的鉴定1、棉花注射vigs液体约一周后,阳性对照出现白化叶片时,对棉花幼苗进行接菌处理。2、黄萎病菌选用大丽轮枝菌vd080(朱荷琴老师惠赠),在查氏液体培养基中进行暗培养10d,温度25℃、转速150r·min-1。稀释至孢子数2×107个·ml-1。3、待棉花发病,根据棉花的发病情况,初步鉴定目的基因在棉花黄萎病抗性中发挥的作用。含注入含pyl156-pds载体vigs悬浮液的棉花植株在注射一周后真叶逐渐白化,如图14所示,黄萎病菌处理一周后,初显病症,棉花真叶的叶缘开始变黄、变脆,并向下卷曲。发病情况如图所示。图15分别为含pyl156空载体、pyl156-66319的vigs海岛棉3-79植株,图16分别为含pyl156空载体、pyl156-66319的vigs陆地棉tm-1植株。可以看出,pyl156-66319的vigs植株中个别真叶开始显病症,而pyl156空载体植株未发现病症。pyl156-66319的vigs陆地棉tm-1中,几乎所有真叶均出现叶缘变黄卷曲且发脆的病症,而pyl156空载体植株发病真叶相对较少。总体来看,陆地棉tm-1比海岛棉3-79的的发病情况较重。通过构建vigs载体注射棉花子叶后,待阳性对照出现白化性状时开始进行黄萎病菌诱导处理,接菌一周后逐渐开始显病症。从棉花的发病情况可以看出,不论是在海岛棉3-79还是在陆地棉tm-1中,pyl156-66319的vigs植株发病情况均比pyl156空载体vigs植株严重。说明gbdrp66319基因沉默表达后对黄萎病菌抗性减弱,初步验证gbdrp66319基因对棉花黄萎病的抗性有一定的作用。并且还可以明显看出,陆地棉tm-1比海岛棉3-79的发病情况较重,由此可以看出海岛棉3-79对黄萎病的抗性明显高于陆地棉tm-1,并且可以推测,这两类基因在陆地棉中对黄萎病菌的抗性也起着非常重要的作用。上述说明示出并描述了发明的若干优选实施例,但如前所述,应当理解发明并非局限于本文所披露的形式,不应看作是对其他实施例的排除,而可用于各种其他组合、修改和环境,并能够在本文所述发明构想范围内,通过上述教导或相关领域的技术或知识进行改动。而本领域人员所进行的改动和变化不脱离发明的精神和范围,则都应在发明所附权利要求的保护范围内。序列表<110>中国农业科学院<120>一种棉花gbdrp66319基因、编码蛋白及应用<130>2017<160>10<170>patentinversion3.3<210>1<211>879<212>dna<213>海岛棉<400>1atgcttgctacaagagataaaacagtaacaattggaggagtccagaattgggaaggtgag60tcaattatgcacccaattgaaattcaacagttgaatatttcaagttacaactatttgaga120aacttagtcgatgataattcttccttcaaaaaagtgattggcttgagggtttgggtttgt180gaagggatagagtgtgttgtttccctgtcctcttttgcctcttcttccactcatccattt240caaagcctcgaggtgttagatcttggagatctgccaaagttgagtgcccttattatgaaa300gatgcaggaattggttcagcaacaacatcaacatcggctccgtctgccaccttttcccat360cttaaggaaattaggatatacaaatgcccaggtatgaagacgttgcttccacattggttg420cttccaaacctccaaagcctggaagaaatacatgtgggtgcttgtagtcagctagtagaa480atattgggagcagcaacatcaaaagttgaagaaaaagagagtgatgcattaatcaaattc540catctttccaaattgagagtattgagactaaataagttaccaaatttgaagagcatatgc600agcaaaagtggagtgatggtttgtgattctctccaagttatcaaagtttttggagattgt660gataaactgaagagaattcctccatttgttccccttgttggcaatgggcagccatttgta720tatgctccaccttctcttacaatcacgccagacacagaatggtgggaatcgttggagtgg780gatgatcatccaaactttaaaaatgttcttcaacccctttggaagcttttaagttatcca840acttatccaacttatccaagatgctggagactgttataa879<210>2<211>292<212>prt<213>海岛棉<400>2metleualathrargasplysthrvalthrileglyglyvalglnasn151015trpgluglygluserilemethisproilegluileglnglnleuasn202530ilesersertyrasntyrleuargasnleuvalaspaspasnserser354045phelyslysvalileglyleuargvaltrpvalcysgluglyileglu505560cysvalvalserleuserserphealaserserserthrhisprophe65707580glnserleugluvalleuaspleuglyaspleuprolysleuserala859095leuilemetlysaspalaglyileglyseralathrthrserthrser100105110alaproseralathrpheserhisleulysgluileargiletyrlys115120125cysproglymetlysthrleuleuprohistrpleuleuproasnleu130135140glnserleuglugluilehisvalglyalacysserglnleuvalglu145150155160ileleuglyalaalathrserlysvalgluglulysgluseraspala165170175leuilelysphehisleuserlysleuargvalleuargleuasnlys180185190leuproasnleulysserilecysserlysserglyvalmetvalcys195200205aspserleuglnvalilelysvalpheglyaspcysasplysleulys210215220argileproprophevalproleuvalglyasnglyglnpropheval225230235240tyralaproproserleuthrilethrproaspthrglutrptrpglu245250255serleuglutrpaspasphisproasnphelysasnvalleuglnpro260265270leutrplysleuleusertyrprothrtyrprothrtyrproargcys275280285trpargleuleu290<210>3<211>22<212>dna<213>人工序列<400>3agatacccgctggacttttacc22<210>4<211>18<212>dna<213>人工序列<400>4tttcgcaccaatggaggc18<210>5<211>26<212>dna<213>人工序列<400>5ttgtgaagggatagagtgtgttgttt26<210>6<211>20<212>dna<213>人工序列<400>6cagacggagccgatgttgat20<210>7<211>33<212>dna<213>人工序列<400>7ggactctagaggatccttcaccttcagcacttg33<210>8<211>40<212>dna<213>人工序列<400>8gatcggggaaattcgagctcaaatggtactaaaatggaga40<210>9<211>26<212>dna<213>人工序列<400>9cgggatccgatggagccgatgttgat26<210>10<211>26<212>dna<213>人工序列<400>10ccctcgagccagaattgggaaggtga26当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 蝎神经毒素AaIT及其编码基因与应用的制造方法与工艺
- 腈水解酶突变体及其编码基因和应用的制造方法与工艺
- 植物抗旱相关蛋白AsTMK1及其编码基因和应用的制造方法与工艺
- 植物抗旱相关蛋白EtSnRK2.2及其编码基因和应用的制造方法与工艺
- OsSAPK7蛋白及其编码基因在提高水稻白叶枯病抗性中的应用的制造方法与工艺
- 一种抗除草剂抗虫融合基因、编码蛋白及应用的制造方法与工艺
- 毛果杨PtrZFP103基因及其编码蛋白和应用的制造方法与工艺
- 大豆bHLH转录因子基因GmFER及其编码蛋白和应用的制造方法与工艺
- 一种花狭口蛙的基因及其编码的多肽和该多肽的用途的制造方法与工艺
- 一种泽陆蛙基因及其编码的多肽和该多肽的用途的制造方法与工艺
- 降低种子重量的白桦AP2基因及其编码蛋白的制造方法与工艺
- 可编码ArtRD蛋白基因及其蛋白和应用的制造方法与工艺
- 蝎神经毒素AaIT及其编码基因与应用的制造方法与工艺
- 腈水解酶突变体及其编码基因和应用的制造方法与工艺
- 植物抗旱相关蛋白AsTMK1及其编码基因和应用的制造方法与工艺
- 植物抗旱相关蛋白EtSnRK2.2及其编码基因和应用的制造方法与工艺
- OsSAPK7蛋白及其编码基因在提高水稻白叶枯病抗性中的应用的制造方法与工艺
- 一种抗除草剂抗虫融合基因、编码蛋白及应用的制造方法与工艺
- 毛果杨PtrZFP103基因及其编码蛋白和应用的制造方法与工艺
- 大豆bHLH转录因子基因GmFER及其编码蛋白和应用的制造方法与工艺
- 腈水解酶突变体及其编码基因和应用的制造方法与工艺
- 植物抗旱相关蛋白AsTMK1及其编码基因和应用的制造方法与工艺
- 植物抗旱相关蛋白EtSnRK2.2及其编码基因和应用的制造方法与工艺
- OsSAPK7蛋白及其编码基因在提高水稻白叶枯病抗性中的应用的制造方法与工艺
- 一种抗除草剂抗虫融合基因、编码蛋白及应用的制造方法与工艺
- 毛果杨PtrZFP103基因及其编码蛋白和应用的制造方法与工艺
- 大豆bHLH转录因子基因GmFER及其编码蛋白和应用的制造方法与工艺
- 一种花狭口蛙的基因及其编码的多肽和该多肽的用途的制造方法与工艺
- 一种泽陆蛙基因及其编码的多肽和该多肽的用途的制造方法与工艺
- 一种水稻MIT1基因、其编码蛋白及应用的制造方法与工艺
- 植物抗旱相关蛋白EtSnRK2.2及其编码基因和应用的制造方法与工艺
- OsSAPK7蛋白及其编码基因在提高水稻白叶枯病抗性中的应用的制造方法与工艺
- 一种抗除草剂抗虫融合基因、编码蛋白及应用的制造方法与工艺
- 毛果杨PtrZFP103基因及其编码蛋白和应用的制造方法与工艺
- 大豆bHLH转录因子基因GmFER及其编码蛋白和应用的制造方法与工艺
- 一种水稻MIT1基因、其编码蛋白及应用的制造方法与工艺
- 蛋白StTrxF、编码基因及其在提高植物淀粉含量中的应用的制造方法与工艺
- 蛋白质及其编码基因在调控植物抗黄萎病性中的应用的制造方法与工艺
- 甘薯耐逆相关蛋白IbFBA2、编码基因、重组载体与应用的制造方法与工艺
- 植物抗旱相关蛋白ZmNAC111及其编码基因与应用的制造方法与工艺
- 植物抗旱相关蛋白EtSnRK2.2及其编码基因和应用的制造方法与工艺
- OsSAPK7蛋白及其编码基因在提高水稻白叶枯病抗性中的应用的制造方法与工艺
- 一种抗除草剂抗虫融合基因、编码蛋白及应用的制造方法与工艺
- 毛果杨PtrZFP103基因及其编码蛋白和应用的制造方法与工艺
- 大豆bHLH转录因子基因GmFER及其编码蛋白和应用的制造方法与工艺
- 一种水稻MIT1基因、其编码蛋白及应用的制造方法与工艺
- 蛋白StTrxF、编码基因及其在提高植物淀粉含量中的应用的制造方法与工艺
- 植物抗旱相关蛋白ZmNAC111及其编码基因与应用的制造方法与工艺
- 核盘菌凝集素SSL‑6蛋白及其编码基因与应用的制造方法与工艺
- 植物抗病毒相关蛋白GmSN1及其编码基因和应用的制造方法与工艺
- 植物抗旱相关蛋白EtSnRK2.2及其编码基因和应用的制造方法与工艺
- OsSAPK7蛋白及其编码基因在提高水稻白叶枯病抗性中的应用的制造方法与工艺
- 一种抗除草剂抗虫融合基因、编码蛋白及应用的制造方法与工艺
- 毛果杨PtrZFP103基因及其编码蛋白和应用的制造方法与工艺
- 大豆bHLH转录因子基因GmFER及其编码蛋白和应用的制造方法与工艺
- 一种水稻MIT1基因、其编码蛋白及应用的制造方法与工艺
- 蛋白StTrxF、编码基因及其在提高植物淀粉含量中的应用的制造方法与工艺
- 蛋白质及其编码基因在调控植物抗黄萎病性中的应用的制造方法与工艺
- 植物抗旱相关蛋白ZmNAC111及其编码基因与应用的制造方法与工艺
- 核盘菌凝集素SSL‑6蛋白及其编码基因与应用的制造方法与工艺