含CF2的2‑噁唑啉酮类化合物及其制备方法与流程
本发明属于化学合成技术领域,涉及2-噁唑啉酮类化合物的制备技术,具体涉及含cf2的2-噁唑啉酮类化合物及其制备方法。
背景技术:
近年来,化学学科的日益发展和不断完善使得人们逐渐开始尝试利用化学的手段高效地将co2转化为一系列重要的化学化工产品,这一变废为宝的举措必将从一定程度上加速自然界的碳循环,使得资源的利用更符合可持续发展的理念,同时利用co2这种无毒无害、廉价易得且可再生的理想的资源构建一系列具有重要意义的化合物既能促进有机化学的发展又符合绿色化学发展的观念。
温和条件下co2的利用尽管存在着巨大的挑战,但是具有非常重要的研究意义和学术价值。不难发现co2除了可以提供丰富的碳源外,还可以提供丰富的氧源,因此如果能高效的利用其中的氧源我们还能够构建一系列的含杂原子化合物或杂环化合物。而这类化合物往往在医药、农药、材料及有机合成等领域表现出突出的性质。因此利用co2来定向合成一系列高附加值的产品是亟待探索的一个领域。
在过去几十年中,许多的研究都集中在将氟原子或含氟基团特别是含有cf3基团的全氟烷基引入到有机分子中。然而,与全氟烷基化方法相比,部分氟代甲基或易于后续衍生化的官能化氟化基团的引入仍然很少。在这些氟化基团中,官能团化的二氟化基团(cf2-fg)是非常有吸引力的,因为二氟亚甲基(cf2)通常用作氧原子或羰基的生物电子等排体,并且在生物活性分子中起非常重要的作用。
含有cf2片段的化合物的传统合成通常可以通过使用诸如dast和dast的衍生物等有害试剂的脱氧氟化反应或使用氟化原料作为结构单元进行,这些方法由于氟化试剂的不稳定性和官能团兼容性差而受到限制。
此前,发明人所在的课题组通过利用整个“co2”部分调节氮-三氟甲基化至氧-三氟甲基化,成功地在铜催化剂下有效地合成含有cf3的2-噁唑啉酮。南开大学的何良年教授课题组利用同样的策略开发了用co2对烯丙胺进行无金属光引发的羧基环化,合成了含全氟烷基的2-噁唑啉酮,但是底物范围非常有限。虽然如此,合成部分氟烷基化的2-噁唑啉酮类物质仍然值得探索,而且利用多组分反应同时将co2应用于二氟烷基杂环化合物的合成中还未见报道。
技术实现要素:
针对现有技术的缺点和技术需求,本发明的目的之一在于提供含cf2的2-噁唑啉酮类化合物的制备方法,该方法包括如下步骤:
以化学结构式如式<ⅰ>所示的物质作为底物,加入二氟化试剂,在极性溶剂中,以及在有光催化剂和dabco参与的条件下,于co2氛围中,利用蓝光led照射,反应得到所述含cf2的2-噁唑啉酮类化合物,其化学结构式如式<ⅱ>、<ⅲ>或<ⅵ>所示;
其中,r1、r2相互独立的为h或ch3,r4为苄基或正丁基;r3包括
所述二氟化试剂为brcf2cooet、cf2po(oet)2、溴二氟乙酰胺、二氟甲基砜或偕二氟炔丙基溴。
在进行本发明之前,是存在一些挑战和难点的。首先是针对选择性问题——co2的引入必然会导致反应亲核位点产生变化,我们都知道分子内的反应一般快于分子间的反应,所以在此反应中氮-二氟烷基化、氧-二氟烷基化仍然存在竞争过程,且从动力学角度讲氧-二氟烷基化处于劣势。第二是反应区域选择性——形成六元环产物还是五元环产物,这一点相对来说比较好解决——利用电子效应和位阻效应合理设计底物可以较好的解决。第三就是体系的兼容性问题,虽然可见光催化已经发展的相当成熟,但是将其与co2的固定结合起来的研究相对较少,所以究竟将两者融合是否能发生预期的效果还值得探索。
通过大量的实验摸索,发明人巧妙的找到了上述制备方法。进行实验摸索的一些内容如本发明的实施例所示。
所述极性溶剂包括dce、thf、dma、dmf中的至少一种。
优选的,所述极性溶剂为dmf。
所述光催化剂包括ir(ppy)2(dtbbpy)(pf6)、ir(df-cf3-ppy)2(dtbbpy)(pf6)、ru(bpy)3cl2·6h2o中的至少一种。
优选的,所述光催化剂为ru(bpy)3cl2·6h2o。
优选的,所述底物与二氟化试剂的摩尔比为1:0.3。
优选的,所述底物与dabco的当量比为1:2。
优选的,所述二氟化试剂为brcf2cooet。
进行所述反应时,于一个大气压co2的氛围下进行;和/或,进行所述蓝光led照射时,采用30瓦的蓝光led灯;和/或,进行所述反应时,于室温中进行。
优选的,进行所述反应时,反应时间为10小时。
本发明的另外一个目的在于提供由上述方法制备得到的所述含cf2的2-噁唑啉酮类化合物。
本发明的有益效果:
本发明借助光催化的体系将二氧化碳利用和二氟烷基化巧妙地结合起来,在温和反应条件下合成了一系列含cf2的2-噁唑啉酮类化合物,本发明的反应具有很好的官能团兼容性差和广泛的底物范围等优点。在本发明的基础上,有望合成一系列含有重要基团的重要骨架,这必将对药物的研发和应用打下非常坚实的基础。
附图说明
图1~图6为本发明所得部分产品的结构谱图。
具体实施方式
下面通过实施例对本发明进行具体描述,有必要在此指出的是以下实施例只是用于对本发明进行进一步的说明,不能理解为对本发明保护范围的限制,该领域的技术熟练人员根据上述发明内容所做出的一些非本质的改进和调整,仍属于本发明的保护范围。
在下述本发明实施例以及本发明说明书其它地方中主要涉及到的略缩词全称如下:
bhtbutylatedhydroxytoluene,二叔丁基-4-甲基苯酚
bnbenzyl,苄基
bubutyl,丁基
bzbenzoyl,苯酰基
boct-butyloxycarbonyl,叔丁氧羰基
co2carbondioxide,二氧化碳
dastdiethylaminotrifluorosulfur,二乙胺基三氟化硫
dabco1,4-diaza[2.2.2]bicyclooctane,三乙烯二胺
dbn2,3,4,6,7,8-hexahydropyrrolo[1,2-a]pyrimidine,1,5-二氮杂双环[4.3.0]
壬-5-烯
dbu1,5-diaza(5,4,0)undec-5-ene,1,5-二氮杂二环[5.4.0]十一-5-烯
dce1,2-bichloroethane,1,2-二氯乙烷
dman,n-dimethylacetamide,n,n-二甲基乙酰胺
dmfn,n-dimethylformamide,n,n-二甲基甲酰胺
etethyl,乙基
gcgaschromatography,气相色谱仪
gc-msgaschromatography-massspectrometer,气相色谱-质谱联用仪
ledlightemittingdiode,发光二极管
memethyl,甲基
mecnacetonitrile,乙腈
mtbd1-methyl-1,3,4,6,7,8-hexahydro-2h-pyrimido[1,2-a]pyrimidine,7-甲
基-1,5,7-三氮杂二环[4.4.0]癸-5-烯
phphenyl,苯基
tbd1,5,7-triazabicylo[4.4.0]dec-5-ene,1,5,7-三叠氮双环(4.4.0)癸-5-烯
tempo2,2,6,6-tetramethylpiperidinooxy,2,2,6,6-四甲基哌啶氧化物
thftetrahydrofuran,四氢呋喃
tipstriisopropylsilyl,三异丙基硅基
tmstrimethylsilyl,三甲基硅基
tsp-toluenesulfonyl,对甲苯磺酰基
在下述本发明实施例以及本发明说明书其它地方中主要涉及到的略缩词的结构如下:
实施例1
碱的考察
我们初步的探索选用n-苄基-2-苯基-1-丙烯胺(1a)作为模板底物,并使用低成本、被广泛使用的溴二氟乙酸乙酯(brcf2cooet)作为二氟化试剂(表1)。首先,我们简单结合了我们以前的工作和光氧化还原催化产生自由基的过程(表1编号1),我们使用1mol%的fac-ir(ppy)3作为光催化剂,dbu作为碱,mecn做溶剂,在一个大气压co2氛围下用30瓦的蓝光led灯照射10h。不幸的是,没有检测到所需的产品,大部分的1a都保留。但是接下来的结果显示如果没有dbu存在,目标产物2a可以在gc-ms上观察到,这一奇怪的现象证实了我们的设计是有可能实现的,同时也证实了我们以前的猜测——碱可能会影响胺的亲核性或活化二氧化碳的过程。于是我们对其他的各种类型的碱进行了考察(表1)。我们发现,反应中使用csf、cs2co3、k2co3等弱的无机碱时,反应效果并没有得到明显改善,而使用强的无机碱naotbu时几乎不能得到目标产物,而使用这些碱时都有不同程度的氮杂环丙烷类副产物被检测到,我们推测在这些碱的作用下co2并没有很好的参与到反应中去,同时不同的碱对于光催化体系与co2活化体系的兼容可能至关重要,而我们的实验结果也很好的证实了这一点。我们非常高兴的发现当使用dabco参与到我们的反应时,我们能以79%的gc收率得到我们的目标产物,而在此条件下,副产物可以被抑制到5%以内,证明使用dabco可以很好的实现co2的利用和选择性的控制。我们认为,dabco可能对于co2的活化和光催化循环都至关重要。
表1碱的筛查a
a除非特别说明,反应底物1a的投料量为0.2mmol(1.0当量),brcf2cooet(0.3mmol),光催化剂1mol%,反应物浓度0.1mol/l,碱(2.0当量),rt即在室温条件下;b用正十二烷作为内标计算的gc收率。
实施例2
溶剂的考察:
确定了最佳的碱dabco之后,接着我们考察了不同的反应溶剂对该反应的影响(表2)。反应在弱极性溶剂dce和中等极性溶剂thf中都可以顺利地进行,但是收率有明显降低。在强极性溶剂dma中,反应收率有略微降低。但是当以强极性溶剂dmf做溶剂时,目标产物的gc收率可以提高到85%,故dmf是目前为止最佳的反应溶剂。
表2溶剂的筛查a
a除非特别说明,反应底物1a的投料量为0.2mmol(1.0当量),brcf2cooet(0.3mmol),光催化剂1mol%,反应物浓度0.1mol/l,dabco(2.0当量),rt即在室温条件下;b用正十二烷作为内标计算的gc收率。
实施例3
光催化剂的考察
确定了最佳溶剂dmf和最佳碱dabco之后,我们又对常用的光催化剂进行了考察(表3),主要考察了ir(ppy)2(dtbbpy)(pf6)、ir(df-cf3-ppy)2(dtbbpy)(pf6)、ru(bpy)3cl2·6h2o。研究发现不同的光催化剂均能比较高效得到目标产物,值得一提的是ru(bpy)3cl2·6h2o略微显示出一定的优异性,不仅是反应性方面,而且从经济上来考虑也略占优势,因为我们都知道到金属钌的价格要比铱的价格便宜。所以我们最终选择了ru(bpy)3cl2·6h2o作为理想的催化剂。并且高兴地发现,光催化剂的当量可以进一步减少到0.5mol%(表3编号5),在此条件下可以以82%的分离收率得到我们的目标产物,这证明了我们的反应具有高效的特点。
综上,我们通过对碱、溶剂和光催化剂进行筛查,最终确定我们的标准反应条件为表3编号5所示的条件。
表3光催化剂的筛查a
a除非特别说明,反应底物1a的投料量为0.2mmol(1.0当量),brcf2cooet(0.3mmol),光催化剂1mol%,反应物在dmf中的浓度0.1mol/l,dabco(2.0当量),rt即在室温条件下;b用正十二烷作为内标计算的gc收率,括号内为分离收率;c使用0.5mol%的光催化剂。
实施例4
确定了标准条件之后,我们接着通过控制实验(表4)证明了可见光、光催化剂、碱和co2对目标产物的生成都是必不可少的,这与我们之前所设计过程完全相符,从而证实了将可见光催化体系与co2活化结合起来是可行的。
表4控制实验a
a除非特别说明,反应底物1a的投料量为0.2mmol(1.0当量),brcf2cooet(0.3mmol),ru(bpy)3cl2·6h2o0.5mol%,反应物在dmf中的浓度0.1mol/l,dabco(2.0当量),rt即在室温条件下,n.d.表示未检测到;b用正十二烷作为内标计算的gc收率,括号内为分离收率;c没有光;d没有co2。
实施例5
底物扩展
如实施例1~4的记载,通过一系列的条件优化过程,我们最终确立了我们的最优反应条件也就是标准条件如表3编号5所示,在此条件的基础之上,我们进行了一系列底物扩展工作,我们分别考察了不同烯丙胺类底物1和不同二氟烷基化试剂,为探索方法学的底物适应性打下了基础。
如表5所示,以brcf2cooet为二氟烷基化试剂,我们首先对烯丙胺类底物1的范围进行了考察。各种不同烯丙胺类底物都可以顺利地进行反应得到相应的含cf2cooet基团的2-噁唑啉酮类化合物,产率从中等到良好不等。除了氮上是苄基外,正丁基也可以产生与之相当产率的产物2ba,而氮上为苯基只有痕量产物2bb被检测到。不幸的是,一些常见的吸电子基团如bz,boc,ts和游离胺不能产生相应的产物,这可能是由于氮的较弱的亲核性。乙烯基芳环上不论是吸电子取代基和给电子取代基均能顺利地得到相应产物。许多官能团,如氟(2c),氯(2d和2r),溴(2e和2t),酯(2f,2g和2n),醚(2h,2k和2p),酰胺(2q)在反应中显示出良好的相容性,这对下游产品的转化非常有益。空间位阻更大的的内部烯烃1u也能以1:1比例的非对映异构体高产率地得到目标产物。不同位置取代的萘环类烯丙胺衍生物(1v和1w)也可以顺利地发生反应。鉴于螺杂环的重要性,在这里我们也成功的实现了一例螺环化合物的合成,显示了良好的产率和高非对映选择性(2x)。值得注意的是,通过改变底物氮和双键之间的距离,我们以中等收率得到的六元环的二氟代烷基化羧基环化产物2y,从而证明了该体系的优越性。另外值得一提的是,苯基在烯烃末端的底物1z也能成功实现转化。
为了进一步扩大该方法的底物范围,我们有考察了各种二氟烷基试剂(表6)。当在标准反应条件下检测溴二氟乙酰胺3a时,得到良好的收率。有趣的是,在该反应中成功地应用了tips保护的偕二氟炔丙基溴化物3b,这在光氧化还原催化剂体系中很少被报道为cf2自由基前体,这给更多的转化提供了极大可能性。以54%的总产率分别获得了保护和脱保护的产物4ba和4bb。值得注意的是,由于膦酰二氟甲基cf2po(oet)2在药物化学中是重要的部分,所以成功应用该方法合成4c显示出强大的潜力。此外,通过使用cf2h基自由基前体5a代替brcf2cooet作为二氟烷基自由基前体有效的合成了含cf2h的2-噁唑啉酮6a。
总的来讲,通过对不同的烯丙胺类化合物和不同的二氟烷基化试剂的考察,我们合成了一系列含cf2的2-噁唑啉酮化合物,这些实验结果充分展示了我们的反应体系具有较广阔的底物适用范围。
表5烯丙胺类底物范围a
a标准条件,所有产率均为分离收率。b非对映异构体比例d.r.=1:1;cd.r.=10:1;dd.r.>19;n.d.表示未检测到。
表6二氟烷基试剂的范围a
a标准条件,所有产率均为分离收率;b36h;c使用2mol%的fac-ir(ppy)3作为催化剂,72h。
实施例6
为了更好地完善我们的方法,我们对目标产物2a进行了克级规模化生产(表7式a)和衍生化(表7式a)。结果显示克级反应仍然可以以相当产率84%高效的得到目标产物,这为产物的后续工业化应用提供了可能。另外,使用氢氧化锂我们可以选择性的水解与二氟烷基相连的酯基得到10,利用不同的还原剂nabh4和lialh4我们可以高效、高选择性地得到部分还原和全部还原的产物。
表7
实施例7
本实施例对上述实施例1~6中的合成过程,以其中一种合成方法为例,说明合成过程的具体步骤:
将装有磁子的10mlschlenk反应管在真空下用热风枪加热高温除水,并在氮气氛围下逐渐恢复至室温。然后在空气中加入0.5mol%ru(bpy)3cl2·6h2o和固体底物(0.20mmol),然后将反应管转移进手套箱内称量dabco(0.40mmol),称量完后将反应管拿出手套箱,置于n2氛围下,接着在n2氛围下用移液枪依次加入液体烯丙胺衍生物(0.20mmol),brcf2co2et(0.30mmol,1.5equiv),无水dmf(2ml)。加完后,将混合物采取液氮冷冻脱气(3次)的方式除掉溶剂里的溶解氧(注意co2会被液氮冷冻造成危险)。然后向反应管中充入co2并在一个大气压的co2氛围下拧紧反应管,接着将反应管置于25度的水浴锅中并用30瓦蓝色led灯照射使其室温反应10小时。反应完后,旋干反应液,柱层析分离纯化(pe:ea10:1~5:1)得目标产物。
对于上述实施例中所提到的一些反应物,发明人将提供相关的合成方法。但这不作为本发明的限制。
底物1a的合成
由于1a比较容易制备,所以本文中我们选用了1a作为我们的模板底物。其合成方法如下。
实验步骤:
在250ml带磁子的茄形瓶中依次加入12.6gn-溴代丁二酰亚胺(nbs),1.02g对甲苯磺酸后,将160mlthf用注射器注入,然后放置于100度的油浴锅上充分搅拌。最后用注射器加入7ga,反应过夜。tlc监测原料大部分消失后,停止反应,待冷至室温后加入少量石油醚,水洗三次,有机相用无水硫酸钠干燥后旋干溶剂,柱层析分离,得4.5gb的粗品为黄色油状物,直接将其用于下一步反应。取化合物b约4g溶于150ml乙腈中,然后回流搅拌条件下依次加入3g碳酸钾,6.6g苄胺,反应12h。tlc监控至原料基本消失后,待冷至室温后过滤,乙酸乙酯洗涤,旋蒸除去溶剂后柱层析分离得到纯品浅黄色油状液体约2.3g。
产品表征数据:1hnmr(400mhz,cdcl3)δ7.47–7.39(m,2h),7.37–7.19(m,8h),5.46–5.39(m,1h),5.26(d,j=1.3hz,1h),3.80(s,2h),3.67(d,j=0.6hz,2h).
溴二氟乙酰胺3a的合成
实验步骤:
n2氛围下,将5.4mla加入到预先干燥的带磁子的圆底烧瓶中,然后向其中逐滴滴入5.1ml二乙胺,滴加完后继续在n2氛围下室温搅拌过夜。后处理:乙醚稀释反应液,2mol/l的盐酸溶液洗涤,饱和碳酸氢钠和饱和氯化钠溶液依次洗涤,有机相旋蒸除去溶剂后柱层析分离的产品7.4g,浅黄色油状液体。
产品3a表征数据:1hnmr(400mhz,cdcl3)δ3.53(q,j=7.1hz,2h),3.43(q,j=7.1hz,2h),1.25(t,j=7.1hz,3h),1.20(t,j=7.1hz,3h).19fnmr(376mhz,cdcl3)δ-54.43(s).
偕二氟炔丙基溴3b的合成
实验步骤:
n2氛围下,将3.1mla溶于30ml无水thf中,然后置于负78度,待温度稳定后于负78度向其中逐滴滴入6.16ml2.5mol/l的正丁基锂正己烷溶液,滴加完后于负78度继续搅拌约半小时,然后迅速用注射器将冷的呈液体状态的二氟二溴甲烷1.9ml加入其中。然后逐渐升至室温继续搅拌16h。后处理:用饱和氯化氨溶液洗涤,水相用正己烷萃取,合并有机相用无水硫酸钠干燥,过滤后除去溶剂,然后减压蒸馏的目标产物3b呈无色油状液体,2.3g。
产品3a表征数据:1hnmr(400mhz,cdcl3)δ1.10–1.15(m,21h).19fnmr(376mhz,cdcl3)δ-32.35(s).
二氟甲基砜6a的合成
实验步骤:
步骤1(合成b):n2氛围下,将1.1当量3.2gnah加入到预先干燥的n2抽充三次的反应瓶中,然后向其中注入15ml无水dmf,然后将a的dmf溶液逐滴加入到反应瓶中,然后在室温下搅拌约半小时后向反应体系中鼓入一氯二氟甲烷气体约2h,最后将反应瓶置于60度油浴锅中搅拌12h。后处理:加水淬灭,乙醚萃取,有机相用5%氢氧化钾洗涤后,用无水硫酸镁干燥,旋干后进行减压蒸馏,得到13.25g目标产物b为浅黄色油状液体。
步骤2(合成6a):
在一个圆底烧瓶中依次加入b,乙腈,四氯化碳,水和水合氯化钌。将其置于搅拌器上剧烈搅拌,然后向其中加入2.5当量高碘酸钠,最后让其室温下继续反应48h。后处理:饱和碳酸氢钠溶液中和反应液,过滤,滤渣用乙酸乙酯洗涤,滤液用乙酸乙酯萃取三次,合并有机相后用无水硫酸镁干燥,旋干溶剂即得目标产物呈白色固体。
产品3a表征数据:1hnmr(400mhz,cdcl3)δ8.37–8.31(m,1h),8.12–8.06(m,1h),7.76–7.66(m,2h),6.59(t,j=53.1hz,1h).19fnmr(376mhz,cdcl3)δ-121.38(s).
实验1
为了进一步了解反应机理,我们进行了一系列实验研究——包括自由基抑制实验和自由基开环实验。
自由基抑制实验——当我们在标准反应条件下向反应体系中加入1.5当量的自由基抑制剂tempo或bht,不能得到目标产物或只能得到痕量的目标产物,同时我们能够检测到tempo或bht与cf2cooet的加合物,这表明反应过程可能是通过cf2cooet自由基进行的。(表8式a、式b)
自由基开环实验——通过使用1-环丙基-1-苯基乙烯7作为自由基探测试剂进行自由基开环实验,目标产物的2a产率有所下降,同时得到了自由基开环产物8和形式上c-h二氟烷基化产物9的混合物,比例大致为1:1,总收率16%。上述结果也表明反应过程中产生了cf2cooet自由基。(表8式c)
表8机理探索
基于自由基抑制试验和自由基开环实验的结果,我们推测反应是经历cf2cooet自由基的过程进行的。以模板底物为例,我们提出了如下反应可能的机理。
在可见光的激发下二价的钌催化剂由基态变为激发态的二价钌,激发态的二价钌进而被dabco(epox=+0.69vvssce,e[ru(ii)*/ru(i)]=+0.77v)淬灭得到ru(i),ru(i)与二氟烷基化试剂brcf2cooet发生单电子转移得到cf2cooet自由基。同时底物1a与co2在dabco存在下相互作用产生氨基甲酸负离子a,然后a与cf2cooet自由基发生自由基加成反应得到中间体b。接着b被氧化得到碳正离子c,最后中间体c发生分子内的环化反应得到目标产物2a。还要注意的是,目前brcf2co2et和b之间的链传递过程也不能被排除。
基于二氧化碳利用和二氟烷基化的重要意义,发明人借助光催化的体系将二者巧妙地结合起来,温和反应条件下合成了一系列含cf2的2-噁唑啉酮类化合物,该反应具有很好的官能团兼容性差和广泛的底物范围等优点。发明人还对各种不同取代的烯丙胺类化合物化合物进行了考察,结果发现该反应均能顺利进行,同时使用环状烯烃也能够得到螺环化合物。接着发明人也考察了不同种类的二氟烷基化试剂,发现借助光催化体系几类常用的二氟烷基化试剂均能很好的发生反应。初步的机理探索实验显示该反应很可能是通过二氟烷基自由基来进行的。
附注:在上述实施例和实验中,所进行的检测、所用的仪器和试剂如下:
核磁和高分辨质谱均在四川大学化学学院分析测试中心完成:
核磁(1hnmr,13cnmr):brukeradvance-400核磁共0振仪(0.5%tms为内标)
高分辨质谱shimadzulcms-it-tof
气相、气相质谱联用、电喷雾质谱在本实验室完成:
气相-质谱联用仪(gc-ms):agilenttechnologies7890b/gc-systemand5977a/msd
气相色谱仪仪(gc):agilenttechnologies7890b
电喷雾质谱(esi-ms):thermo-itq
反应用薄层色谱(tlc)、气相色谱(gc)、电喷雾质谱(esi-ms)或气质联用色谱(gc-ms)进行监测。展开后的硅胶板在254nm波长的紫外光下直接观察,或置于碘缸中片刻后取出直接观察。
除非特别指出,本论文实验中所用的试剂均为购买后未经任何处理直接使用。所用试剂和药品均为分析纯或化学纯,大部分试剂购自alfaaesar、百灵威、伊诺凯、aldrich、adamas、成都瑞欧克、科龙等公司。无水试剂均来自实验室溶剂处理系统或购买自百灵威、伊诺凯、华威锐科等试剂公司。
柱层析硅胶(200-300目):青岛海洋化工厂生产,未活化。
本领域技术人员可以根据适合的文献程序合成溴二氟乙酰胺3a,偕二氟炔丙基溴3b,二氟甲基砜6a,烯丙基胺类底物1,1-环丙基-1-苯基乙烯7。
部分产品数据表征:
3-benzyl-5-phenyl-5-(2,2-difluoro-3-ethoxy-3-oxopropyl)oxazolidin-2-one(2a)
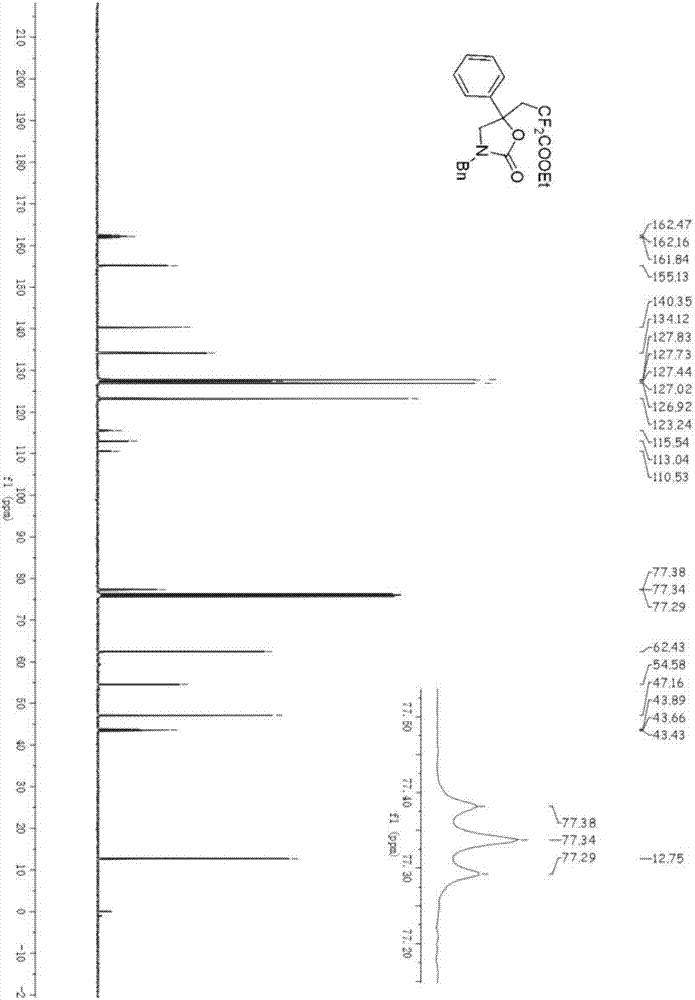
2h),3.64(dd,j=103.6,9.0hz,2h),2.85(t,j=14.2hz,2h),1.33(t,j=7.2hz,3h).;13cnmr(101mhz,cdcl3)δ162.16(t,j=31.6hz),155.13,140.35,134.12,127.83,127.73,127.44,127.02,126.92,123.24,113.04(t,j=252.0hz),77.34(t,j=4.5hz),62.43,54.58,47.16,43.66(t,j=23.2hz),12.75;19fnmr(376mhz,cdcl3)δ-101.25(d,j=270.1hz),-102.08(d,j=270.1hz).exactmassesi-ms:calculatedm/zforc21h21f2nnao4+[m+na]+:412.1331,found:412.1331.
3-butyl-5-phenyl-5-(2,2-difluoro-3-ethoxy-3-oxopropyl)oxazolidin-2-one(2ba)
1hnmr(400mhz,cdcl3)δ7.53–7.29(m,5h),4.34–4.12(m,2h),3.81(dd,j=123.6,8.9hz,2h),3.24(dtd,j=21.5,14.1,7.6hz,2h),2.90(t,j=14.5hz,2h),1.56–1.42(m,2h),1.32(t,j=7.2hz,3h),1.37–1.20(m,2h),0.90(t,j=7.3hz,3h);13cnmr(101mhz,cdcl3)δ163.19(t,j=31.5hz),156.04,141.72,128.79,128.44,124.28,114.18(t,j=251.8hz),78.02(t,j=4.5hz),63.46,56.10,44.81(t,j=23.1hz),43.78,29.20,19.70,13.77,13.65;19fnmr(376mhz,cdcl3)δ-101.13(d,j=269.6hz),-102.20(d,j=269.6hz).exactmassesi-ms:calculatedm/zforc18h24f2no4+[m+h]+:356.1668,found:356.1666,c18h23f2nnao4+[m+na]+:378.1487,found:378.1475.
3-benzyl-5-(4-fluorophenyl)-5-(2,2-difluoro-3-ethoxy-3-oxopropyl)oxazolidin-2-one(2c)
2h),4.33–4.18(m,2h),3.62(dd,j=107.4,9.1hz,2h),2.95–2.74(m,2h),1.34(t,j=7.2hz,3h);13cnmr(101mhz,cdcl3)δ162.11(t,j=31.6hz),161.46(d,j=248.1hz),154.94,136.07(d,j=3.2hz),134.03,127.88,127.11,126.92,125.29(d,j=8.3hz),114.68(d,j=21.8hz),112.93(t,j=252.1hz),77.06(t,j=4.4hz),62.51,54.70,47.17,43.68(t,j=23.1hz),12.75;19fnmr(376mhz,cdcl3)δ-101.34(d,j=270.3hz),-102.12(d,j=270.3hz),-113.19.exactmassesi-ms:calculatedm/zforc21h21f3no4+[m+h]+:408.1417,found:408.1408.
3-benzyl-5-(4-chlorophenyl)-5-(2,2-difluoro-3-ethoxy-3-oxopropyl)oxazolidin-2-one(2d)
hz,2h),4.33–4.19(m,2h),3.60(dd,j=110.9,9.1hz,2h),2.93–2.74(m,2h),1.34(t,j=7.2hz,3h);13cnmr(101mhz,cdcl3)δ162.10(t,j=31.5hz),154.85,138.78,133.97,133.47,127.93,127.90,127.14,126.93,124.83,112.88(t,j=252.3hz),76.97(t,j=4.4hz),62.55,54.60,47.21,43.57(t,j=23.2hz),12.76;19fnmr(376mhz,cdcl3)δ-101.33(d,j=270.5hz),-102.09(d,j=270.5hz).exactmassesi-ms:calculatedm/zforc21h20clf2nnao4+[m+na]+:446.0941,found:446.0945.
3-benzyl-5-(4-bromophenyl)-5-(2,2-difluoro-3-ethoxy-3-oxopropyl)oxazolidin-2-one(2e)
4.33–4.19(m,2h),3.60(dd,j=111.5,9.1hz,2h),2.92–2.73(m,2h),1.34(t,j=7.2hz,3h);13cnmr(101mhz,cdcl3)δ162.09(t,j=31.5hz),154.83,139.30,133.94,130.89,127.90,127.15,126.93,125.11,121.59,112.87(t,j=252.3hz),77.00(t,j=4.4hz),62.56,54.56,47.20,43.50(t,j=23.3hz),12.76;19fnmr(376mhz,cdcl3)δ-101.33(d,j=270.6hz),-102.08(d,j=270.5hz).exactmassesi-ms:calculatedm/zforc21h20brf2nnao4+[m+na]+:490.0436,found:490.0442.
tert-butyl-4-(3-benzyl-2-oxo-5-(2,2-difluoro-3-ethoxy-3-oxopropyl)oxazolidin-5-yl)benzoate(2f)
2h),4.42(dd,j=58.1,14.9hz,2h),4.36–4.21(m,2h),3.63(dd,j=116.6,9.1hz,2h),3.08–2.72(m,2h),1.59(s,9h),1.35(t,j=7.2hz,3h).;13cnmr(101mhz,cdcl3)δ164.99,163.16(t,j=31.5hz),155.90,145.60,135.01,132.19,129.92,128.94,128.17,127.96,124.22,113.93(t,j=252.3hz),81.44,78.21(t,j=4.4hz),63.59,55.77,48.22,44.54(t,j=23.3hz),28.16,13.80;19fnmr(376mhz,cdcl3)δ-101.26(d,j=269.9hz),-102.17(d,j=270.0hz).exactmassesi-ms:calculatedm/zforc26h29f2nnao6+[m+na]+:512.1855,found:512.1849.
3-benzyl-5-(4-(trifluoromethoxy)phenyl)-5-(2,2-difluoro-3-ethoxy-3-oxopropyl)
oxazolidin-2-one(2h)
2h),3.63(dd,j=113.2,9.1hz,2h),2.99–2.72(m,2h),1.34(t,j=7.2hz,3h);13cnmr(101mhz,cdcl3)δ162.07(t,j=31.5hz),154.82,148.08,138.91,133.96,127.89,127.16,126.93,125.09,120.16,119.34(q,j=257.7hz),112.89(t,j=252.3hz),76.97(t,j=4.4hz),62.53,54.62,47.17,43.53(t,j=23.2hz),12.73;19fnmr(376mhz,cdcl3)δ-57.89,-101.33(d,j=270.9hz),-102.08(d,j=270.9hz).exactmassesi-ms:calculatedm/zforc22h20f5nnao5+[m+na]+:496.1154,found:496.1146.
3-benzyl-5-(p-tolyl)-5-(2,2-difluoro-3-ethoxy-3-oxopropyl)oxazolidin-2-one(2i)
3.62(dd,j=100.2,9.0hz,2h),2.83(t,j=14.4hz,2h),2.34(s,3h),1.32(t,j=7.2hz,3h);13cnmr(101mhz,cdcl3)δ162.19(t,j=31.6hz),155.22,137.39,137.28,134.17,128.34,127.82,127.00,126.94,123.19,113.07(t,j=252.0hz),77.37(t,j=4.5hz),62.40,54.58,47.19,43.72(t,j=23.1hz),20.01,12.74;19fnmr(376mhz,cdcl3)δ-101.29(d,j=269.8hz),-102.07(d,j=269.8hz).exactmassesi-ms:calculatedm/zforc22h24f2no4+[m+]+:404.1668,found:404.1670.
3'-benzyl-2-(2,2-difluoro-3-ethoxy-3-oxopropyl)-3,4-dihydro-2h-spiro[naphthalene-1,5'-oxazolidin]-2'-one(2x)
3-benzyl-5-phenyl-5-(2,2-difluoro-3-(diethylamino)-3-oxopropyl)oxazolidin-2-one(4a)
110.2,9.1hz,2h),3.49–3.31(m,2h),3.34–3.25(m,2h),3.10–2.88(m,2h),1.11(dt,j=14.3,7.1hz,6h);13cnmr(101mhz,cdcl3)δ161.14(t,j=28.2hz),155.80,141.57,134.31,127.74,127.57,127.09,126.99,126.89,123.38,117.05(t,j=257.9hz),78.30,54.79,47.13,43.15(t,j=22.0hz),40.92(t,j=6.3hz),40.86,13.18,11.20;19fnmr(376mhz,cdcl3)δ-96.29(d,j=285.4hz),-98.37(d,j=285.4hz).exactmassesi-ms:calculatedm/zforc23h27f2n2o3+[m+h]+:417.1984,found:417.1984;calculatedm/zforc23h26f2n2nao3+[m+na]+:439.1803,found:439.1802.
3-benzyl-5-phenyl-5-(2,2-difluoro-2-(diethoxyphosphoryl)ethyl)oxazolidin-2-one(4c)
104.6,9.1hz,2h),2.91–2.68(m,2h),1.34(dt,j=13.9,7.1hz,6h);13cnmr(101mhz,cdcl3)δ156.68,142.23,135.28,128.81,128.70,128.35,128.05,127.98,124.43,119.49(dd,j=479.4,263.1hz),79.41(d,j=9.7hz),64.96(dd,j=11.0,7.0hz),55.13,48.27,43.24(td,j=19.0,14.9hz),16.39(dd,j=5.3,3.4hz);19fnmr(376mhz,cdcl3)δ-110.57(dd,j=301.2,104.3hz),-111.99(dd,j=301.2,103.6hz).exactmassesi-ms:calculatedm/zforc22h27f2no5p+[m+h]+:454.1590,found:454.1587;calculatedm/zforc22h26f2nnao5p+[m+na]+:476.1409,found:476.1408.
3-benzyl-5-phenyl-5-(2,2-difluoroethyl)oxazolidin-2-one(6a)
hz,2h),3.61(dd,j=57.7,9.0hz,2h),2.52(td,j=15.3,4.7hz,2h);13cnmr(101mhz,cdcl3)δ156.61,141.13,135.14,129.04,128.94,128.58,128.16,128.03,124.12,114.49(t,j=239.6hz),78.91(t,j=6.0hz),55.73,48.27,45.16(t,j=21.8hz);19fnmr(376mhz,cdcl3)δ-113.61(d,j=293.2hz),-114.44(d,j=293.2hz).exactmassesi-ms:calculatedm/zforc18h18f2no2+[m+h]+:318.1300,found:318.1285;calculatedm/zforc18h17f2nnao2+[m+na]+:340.1119,found:340.1098。
- 还没有人留言评论。精彩留言会获得点赞!