一种含苯基三氮唑的补骨脂素衍生物和用途的制作方法
本发明涉及一种含苯基三氮唑的补骨脂素衍生物和用途,该类化合物经过抗白癜风活性筛选,化合物8a、8j、8m-8o共5个含苯基三氮唑的补骨脂素衍生物在制备治疗白癜风的药物中的用途。
背景技术:
白癜风是一种常见的自发或特发性的色素脱失性皮肤病,被称为世界皮肤病三大顽症之一,困扰全世界5000万以上的患者。在世界不同地域、不同种族间发病率从0.1%-8%不等,我国一般人群患病率为0.56%左右,大约一半患者于20周岁以前发病,男性和女性的患病率相等。白癜风主要表现为皮肤、粘膜的白斑及灰/白色毛发等。西医认为白癜风是由于皮肤和毛囊的黑素细胞内酪氨酸酶系统的功能减退、丧失而引起的。
补骨脂为豆科一年生草本植物补骨脂(psoraleacorylifolial.)的干燥成熟果实,收载于《中国药典》、《维吾尔药志》、《中华本草-维吾尔分卷》等,是历代传统维吾尔医治疗白癜风的主要经典药物。维吾尔医学认为白癜风主要由异常粘液质引起,治疗时主张清除异常体液,纠正异常气质,从而恢复机体自然力。补骨脂具有生干生热、清除异常黏液质、净血解毒、增加色素、杀驱肠虫等功效。此外,补骨脂还具有抗菌、雌激素样作用、抗肿瘤、抗氧化、免疫调节和抗抑郁等多种生物活性。
目前以植物补骨脂为主要成分的药物和制剂,临床上主要用于治疗白癜风:如复方补骨脂颗粒、补骨脂注射液、复方补骨脂酊、驱白巴布斯片、复方卡力孜然酊(维药名维阿露)等。尽管维药治疗白癜风效果显著,优势明显,但是其物质基础研究薄弱,作用机制和体内外代谢过程亦不明晰,严重的制约了其二次开发。
经过多年研究,国内外研究人员已经从补骨脂(psoraleacorylifolial.)中分离出补骨脂素类化合物(呋喃香豆素类化合物)、黄酮类、萜类、以及少量脂类和挥发油。其中补骨脂素类化合物(呋喃香豆素类化合物)作为其中的主要化学成分,研究范围最广。
补骨脂素类化合物(psoralen)是目前临床上治疗白癜风最常用的光敏性药物,但是必须配合日光或长波紫外线(uva)照射治疗,所以此种治疗方法称为puva(psoralen+uva)。常用的有8-甲氧基补骨脂素(8-mop)和5-甲氧补骨脂素(5-mop),后来又人工合成了三甲基补骨脂素(tmp),如结构所示。在白癜风的治疗中,puva能够激活酪氨酸酶活性,催化黑素合成,促进黑素细胞的分裂及移动,最终使黑色素合成增加,白斑颜色逐渐恢复。
经检索,有关文献为:
[1]krugerc.,schallreuterk.u.areviewoftheworldwideprevalenceofvitiligoinchildren/adolescentsandadults[j].int.j.dermatol.,2012,51,1206-1212.
[2]王晓艳.中国六省市白癜风流行病学调查[m].中华皮肤科杂志,2010,463-466.
[3]alikhana.,felstenl.m.,dalym.,petronic-rosicv.vitiligo:acomprehensiveoverviewparti.introduction,epidemiology,qualityoflife,diagnosis,differentialdiagnosis,associations,histopathology,etiology,andwork-up[j].j.am.acad.dermatol.,2011,65,473-491.
[4]arcos-burgosm.,parodie.,salgarm.,bedoyae.,builesj.,jaramillod.,ceballosg.,uribea.,riveran.,riverad.,fonsecai.,camargom.,palaciog.vitiligo:complexsegregationandlinkagedisequilibriumanalyseswithrespecttomicrosatellitelocispanningthehla[j].hum.genet.,2002,110,334-342.
[5]alain
[6]hirschhornj.n.,lohmuellerk.,byrnee.,hirschhornk.acomprehensivereviewofgeneticassociationstudies[j].genet.med.,2002,4,45-61.
[7]schallreuterk.u.,krügerc.,rokosh.,hasses.,zothnerc.,panskea.basicresearchconfirmscoexistenceofacquiredblaschkolinearvitiligoandacrofacialvitiligo[j].arch.dermatol.res.,2007,299,225-230.
[8]ezzedinek.,eleftheriadouv.,whittonm.,vangeeln.vitiligo.lancet[j].2015,inpress,doi:10.1016/s0140-6736(14)60763-7.
[9]吐尔逊·乌甫尔,斯拉甫·艾白,热甫哈提·赛买提,居来提·阿不都瓦衣提,伊力卡尔·拜克提亚.维药成熟剂和清除剂治疗166例进展期白癜风疗效观察[j].中药药理与临床,2012,28,161-162.
[10]夏木西努尔·肖盖提,斯拉甫·艾白,热娜·卡斯木.白癜风的药物治疗现状[j].中国民族医药杂志,2011,8,62-64.
[11]黄建,赵陆华,邹巧根.补骨脂化学成分及药理研究进展[j].药学进展,2000,24,212-214.
[12]yins.,fanc.q.,wangy.,dongl.,yuej.m.antibacterialprenylflavonederivativesfrompsoraleacorylifoliaandtheirstructure-activityrelationshipstudy[j].bioorg.med.chem.,2004,12,4387-4392.
[13]zhangc.z.,wangs.x.,zhangy.,chenj.p.,liangx.m.invitroestrogenicactivitiesofchinesemedicinalplantstraditionallyusedforthemanagementofmenopausalsymptoms[j].j.ethnopharmacol.,2005,98,295-300.
[14]bapatk.,chintalwarg.j.,pandeyu.,thakurv.s.,sarmah.d.,samuelg.,pillaim.r,chattopadhyays.,venkateshm.preparationandinvitroevaluationofradioiodinatedbakuchiolasanantitumoragent[j].appl.radiat.isot.,2005,62,389-393.
[15]guoj.n.,wenx.c.,wuh.,liq.h.,bik.antioxidantsfromachinesemedicinalherb-psoraleacorylifolia[j].foodchem.,2005,91,287-292.
[16]lathap.g.,evansd.a.,panikkark.r.,jayavardhanank.k.immunomodulatoryandantitumourpropertiesofpsoraleacorylifoliaseeds[j].fitoterapia,2000,71,223-231.
[17]cheny.,wangh.d.,xiax.,kungh.f.,pany.,kongl.d.behavioralandbiochemicalstudiesoftotalfurocoumarinsfromseedsofpsoraleacorylifoliainthechronicmildstressmodelofdepressioninmice[j].phytomedicine.2007,14,523-529.
[18]yuef.w.,yan.l.,wenx.,dongm.y.,yanz.,xium.g.,yant.x.,aid.q.auplc–ms/msmethodforinvivoandinvitropharmacokineticstudiesofpsoralenoside,isopsoralenoside,psoralenandisopsoralenfrompsoraleacorylifoliaextract[j].j.ethnopharmacol.,2014,151,609-617.
[19]ruanb.,kongl.y.,takayay.,niwam.studiesonthechemicalconstituentsofpsoraleacorylifolial[j].j.asiannat.prod.res.,2007,9,41-44.
[20]hsuy.t.,wuc.j.,chenj.m.,yangy.c.,wangs.y.thepresenceofthreeisoflavonoidcompoundsinpsoraleacorylifolia[j].j.chromatogr.sci.,2001,39,441-444.
[21]amitn.s.,nutanm.,devanandp.,fulzele.determinationofisoflavonecontentandantioxidantactivityinpsoraleacorylifolial.calluscultures[j].foodchem.,2010,118,128-132.
[22]chengz.w.,caix.f.,datn.t.,hongs.s.,hana.r.,seoe.k.,hwangb.y.,nanj.x.,leed.h.,leej.j.bisbakuchiolsaandb,noveldimericmeroterpenoidsfrompsoraleacorylifolia[j].tetrahedronlett.,2007,48,8861-8864.
[23]njoom.d.,westerhofw.vitiligo.pathogenesisandtreament[j].am.j.clin.dermatol.,2001,2,167-181。
本发明在国内外有关专利、文献的综合分析和本课题组前期研究工作的基础上,对该植物中的活性化合物—补骨脂素进行进一步的改造和修饰,通过点击化学反应将不同的苯氧乙炔引入到补骨脂素分子中,提高其成药性,并研究了这些含苯基三氮唑的补骨脂素衍生物对小鼠b16细胞中黑素生成及酪氨酸酶活性的影响,其结果表明了疗效显著、靶点明确、作用机理清晰的抗白癜风新药。
技术实现要素:
本发明的目的在于,提供一种含苯基三氮唑的补骨脂素衍生物和用途,该类衍生物以间苯二酚为起始原料,在硫酸的作用下,首先与乙酰乙酸乙酯缩合得到4-甲基-7-羟基香豆素(中间体1);将中间体1再与α-溴代苯乙酮反应得到4-甲基-7-(2-氧代)苯乙氧基香豆素(中间体2);将中间体2在氢氧化钾乙醇溶液中关环形成5-甲基-3-苯基补骨脂素(中间体3);将中间体3被二氧化硒氧化得到相应的醛,即7-氧代-3-苯基-7h-呋喃[3,2-g]苯并吡喃-5-甲醛(中间体4);再利用硼氢化钠将中间体4还原成5-羟甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(中间体5);中间体5被进一步溴代得到5-溴甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(中间体6);中间体6与叠氮化钠在乙腈中回流得到5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(中间体7);最后,中间体7与不同的取代苯氧乙炔反应得到含苯基三氮唑的补骨脂素衍生物8a-8o。并将得到的15个含苯基三氮唑的补骨脂素衍生物(8a-8o)进行了小鼠b16细胞中黑素生成及酪氨酸酶活性的作用试验,其结果显示,化合物8a、8j、8m-8o共5个含苯基三氮唑的补骨脂素衍生物均可用于临床上制备治疗白癜风的药物中的用途。
本发明所述的一种含苯基三氮唑的补骨脂素衍生物,该类衍生物结构为:
其中:
化合物8a为5-((4-((4-氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8b为5-((4-((2-氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8c为5-((4-((3-氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8d为5-((4-((2,4-二氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8e为5-((4-((3,5-二氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8f为5-((4-((3,4-二氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8g为5-((4-((4-氟苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8h为5-((4-((2-氟苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8i为5-((4-((3,5-二氟苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8j为5-((4-((4-溴苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8k为5-((4-(苯氧基甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8l为5-((4-((2-甲基苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8m为5-((4-((4-甲基苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8n为5-((4-((2-甲氧基苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8o为5-((4-((4-甲氧基苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
所述补骨脂素胺类衍生物中,化合物8a、8j、8m-8o共5个含苯基三氮唑的补骨脂素衍生物均可用于临床上制备治疗白癜风的药物。
本发明所述的一种含苯基三氮唑的补骨脂素衍生物,其结构如通式(i)所示:
本发明所述的一种含苯基三氮唑的补骨脂素衍生物,将得到的15个含苯基三氮唑的补骨脂素衍生物(8a-8o)进行了小鼠b16细胞中黑素生成及酪氨酸酶活性的作用试验,促进黑素合成的结果显示:与阴性对照相比,所有15个衍生物,除8i外均能够促进b16细胞中黑素的生成,且促进作用从102.13%-211.36%不等;与阳性对照相比,化合物8a、8j、8m-8o共5个化合物对黑素生成的促进作用均优于阳性对照,其中化合物8o对黑素生成的促进作用是阳性对照的1.6倍以上;
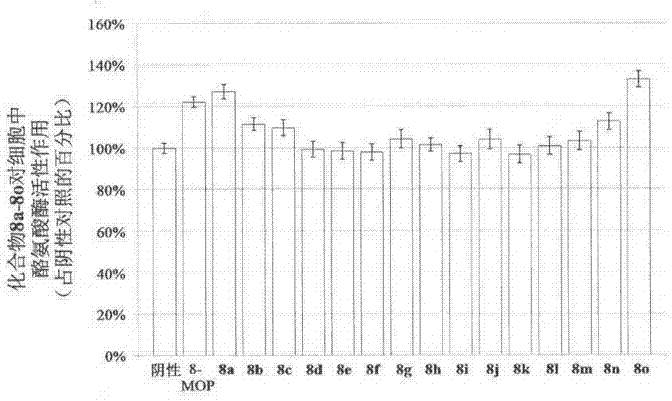
激活酪氨酸酶活性的结果显示:与阴性对照相比,除8d、8e、8f、8i和8k以外的所有化合物均能够激活酪氨酸酶的活性,且激活作用从100.72%-132.80%不等;与阳性对照相比,化合物8a和8o共2个化合物对酪氨酸酶活性的激活作用均优于阳性对照。
综上所述,化合物8a、8j、8m-8o共5个补骨脂素胺类衍生物均可用于临床上制备治疗白癜风的药物中的用途。
本发明所述的一种含苯基三氮唑的补骨脂素衍生物和用途,其中含苯基三氮唑的补骨脂衍生物的制备方法,按下列步骤进行:
中间体1的制备:
a、在冰浴条件下,将间苯二酚溶解在适量的干燥1,4-二氧六环中,搅拌至全部溶解,缓慢滴加浓硫酸,使其温度不超过0℃,滴加完毕后,再加入乙酰乙酸乙酯,然后升温至60℃,充分搅拌使其反应完全,反应液倾倒入冰水中,静置后采用乙酸乙酯萃取,有机相合并干燥,浓缩后得中间体1,4-甲基-7-羟基香豆素;
中间体2的制备:
b、将适量碳酸钾和中间体1溶解在20ml的丙酮中,并加入α-溴代苯乙酮,回流反应至原料全部消失,反应液过滤,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比5∶1的石油醚∶乙酸乙酯,即得中间体2,4-甲基-7-(2-氧代)苯乙氧基香豆素;
中间体3的制备:
c、将步骤b得到的中间体2溶解在无水乙醇中,加入浓度为4%氢氧化钾的乙醇溶液,加热至回流反应,待原料全部消失后,减压浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比10:1的石油醚∶乙酸乙酯,即得中间体3,5-甲基-3-苯基补骨脂素;
中间体4的制备:
d、将步骤c所得到的中间体3和二氧化硒溶解在干燥二甲苯中,加热至回流反应,待原料全部消失后,冷却至室温,母液过滤,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比10:1的石油醚∶乙酸乙酯,即得中间体4,7-氧代-3-苯基-7h-呋喃[3,2-g]苯并吡喃-5-甲醛;
中间体5的制备:
e、将步骤d中所得到的中间体4溶解在无水乙醇中,充分搅拌使其溶解,分次加入硼氢化钠,继续在室温下反应直到原料全部消失,加入稀盐酸淬灭反应,加水稀释后用乙酸乙酯萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比5:1的石油醚∶乙酸乙酯,即得中间体5,5-羟甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
中间体6的制备:
f、将步骤e中得到的中间体5溶解在无水四氢呋喃中,冰浴下搅拌使其溶解,缓慢滴加三溴化磷的四氢呋喃溶液,滴加完毕后移至室温反应,tlc指示反应结束后,向反应体系中加入蒸馏水,并用碳酸氢钠溶液调节ph至中性,二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比10:1的石油醚∶乙酸乙酯,即得中间体6,5-溴甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
中间体7的制备:
g、将步骤f中得到的中间体6和叠氮化钠溶解在无水乙腈中,热至回流反应,待原料全部消失后,冷却至室温,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比10:1的石油醚∶乙酸乙酯,即得中间体7,5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
化合物8a-8o的制备:
h、将步骤g中得到的中间体7和不同的取代苯氧乙炔溶解在体积v叔丁醇∶v水=3:1的混合溶剂中,再加入五水硫酸铜和铜粉,在80℃下反应过夜。待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2:1的石油醚∶乙酸乙酯,即得相应的衍生物8a-8o。
化学反应式为:
(ⅰ)乙酰乙酸乙酯,浓硫酸,温度0-60℃;(ⅱ)α-溴代苯乙酮,碳酸钾,丙酮,回流;(ⅲ)4%氢氧化钾乙醇溶液,回流;(ⅳ)二氧化硒,二甲苯,回流;(ⅴ)硼氢化钠,甲醇,室温;(ⅵ)三溴化磷,四氢呋喃,0℃-rt;(ⅶ)叠氮化钠,乙腈,回流;(ⅷ)不同的苯氧丙炔,五水硫酸铜,铜粉,体积比1:3的叔丁醇:水,温度80℃;
其中r1分别为4-氯、2-氯、3-氯、2.4-二氯、3.5-二氯、3,4-二氯、4-氟、2-氟、3,5-二氟、4-溴、氢、2-甲基、4-甲基、2-甲氧基、4-甲氧基。
附图说明
图1为本发明中含苯基三氮唑的补骨脂素衍生物8a-8o对b16细胞中黑素合成作用图;
图2为本发明中含苯基三氮唑的补骨脂素衍生物8a-8o对b16细胞中酪氨酸酶活性的作用图。
具体实施方式
依据实施例对本发明进一步说明,但本发明不仅限于这些实施例;
试剂:所有试剂均为市售的分析纯;
实施例1
中间体1的制备:
在冰浴条件下,将2.0g间苯二酚溶解在50ml的干燥1,4-二氧六环中,搅拌至全部溶解,缓慢滴加0.5ml浓硫酸,使其温度保持在0℃,滴加完毕后,再加入2.8g乙酰乙酸乙酯,然后升温至60℃,充分搅拌使其反应完全,反应液倾倒入冰水中,静置后采用乙酸乙酯萃取,有机相合并干燥,浓缩后得3.02g中间体1为4-甲基-7-羟基香豆素;
中间体2的制备:
将1.4g碳酸钾和0.88g中间体1溶解在50ml的丙酮中,并加入1.49gα-溴代苯乙酮,回流反应至原料全部消失,反应液过滤,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比5∶1的石油醚∶乙酸乙酯,即得1.25g中间体2为4-甲基-7-(2-氧代)苯乙氧基香豆素;
中间体3的制备:
将2.94g中间体2溶解在100ml无水乙醇中,加入20ml浓度为4%氢氧化钾的乙醇溶液,加热至回流反应,待原料全部消失后,减压浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比10∶1的石油醚∶乙酸乙酯,即得1.34g中间体3为5-甲基-3-苯基补骨脂素;
中间体4的制备:
将5.52g中间体3和3.33g二氧化硒溶解在干燥二甲苯中,加热至回流反应,待原料全部消失后,冷却至室温,母液过滤,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比10∶1的石油醚∶乙酸乙酯,即得3.83g中间体4为7-氧代-3-苯基-7h-呋喃[3,2-g]苯并吡喃-5-甲醛;
中间体5的制备:
将5.80g中间体4溶解在无水乙醇中,充分搅拌使其溶解,分次加入0.38g硼氢化钠,继续在室温下反应直到原料全部消失,加入稀盐酸淬灭反应,加水稀释后用乙酸乙酯萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比5∶1的石油醚∶乙酸乙酯,即得4.56g中间体5为5-羟甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
中间体6的制备:
将2.92g中间体5溶解在25ml的无水四氢呋喃中,冰浴下搅拌使其溶解,缓慢滴加含0.95ml三溴化磷的四氢呋喃溶液,滴加完毕后移至室温反应,tlc指示反应结束后,向反应体系中加入蒸馏水,并用碳酸氢钠溶液调节ph至中性,二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比10∶1的石油醚∶乙酸乙酯,即得3.12g中间体6为5-溴甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮;
中间体7的制备:
将3.55g中间体6和0.96g叠氮化钠溶解在无水乙腈中,热至回流反应,待原料全部消失后,冷却至室温,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比10∶1的石油醚∶乙酸乙酯,即得2.57g中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
实施例2
化合物8a的制备:
将实施例1得到的中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮0.317g和4-氯苯氧乙炔0.199g溶解在体积比3:1的叔丁醇∶水20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在温度80℃下反应过夜,待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.451g化合物8a,名称为5-((4-((4-氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-((4-氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物8a)的核磁数据:
1hnmr(400mhz,cdcl3)δ8.04(s,1h),7.86(s,1h),7.70(s,1h),7.58–7.53(m,5h),7.48–7.41(m,1h),7.21(d,j=9.0hz,2h),6.88(d,j=9.0hz,2h),6.05(s,1h),5.80(s,2h),5.21(s,2h)。
13cnmr(101mhz,cdcl3)δ160.06,157.52,156.70,152.07,148.18,143.47,130.56,129.60,129.54,128.48,127.65,124.70,123.40,122.48,116.24,115.20,114.02,113.62,101.06,62.33,50.95。
实施例3
化合物8b的制备:
将实施例1得到的中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮0.317g和2-氯苯氧乙炔0.199g溶解在体积比3:1的叔丁醇∶水的20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在温度80℃下反应过夜,待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.440g化合物8b,名称为5-((4-((2-氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-((2-氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物8b)的核磁数据:
1hnmr(400mhz,cdcl3)δ8.04(s,1h),7.86(s,1h),7.77(s,1h),7.59–7.50(m,5h),7.48–7.40(m,1h),7.33(d,j=8.0hz,1h),7.22–7.15(m,1h),7.06(d,j=8.2hz,1h),6.94–6.87(m,1h),6.02(s,1h),5.81(s,2h),5.33(s,2h).
13cnmr(101mhz,cdcl3)δ160.05,157.51,153.66,152.08,148.24,145.51,143.42,132.60,131.02,130.57,129.53,128.95,128.46,127.98,127.67,124.68,123.49,122.51,115.16,114.57,113.92,101.05,63.52,50.90。
实施例4
化合物8c的制备:
将实施例1得到的中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮0.317g和-氯苯氧乙炔0.199g3溶解在体积比3:1的叔丁醇∶水的20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在温度80℃下反应过夜,待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.434g化合物8c,名称为5-((4-((3-氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-((3-氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物8c)的核磁数据:
1hnmr(400mhz,cdcl3)δ8.04(s,1h),7.86(s,1h),7.71(s,1h),7.59–7.52(m,5h),7.48–7.41(m,1h),7.22–7.15(m,1h),6.99–6.92(m,2h),6.85(d,j=8.6hz,1h),6.06(s,1h),5.81(s,2h),5.22(s,2h).
13cnmr(101mhz,cdcl3)δ160.06,158.84,157.53,152.09,148.17,143.45,135.14,131.06,130.56,130.50,129.54,128.48,127.67,123.36,121.83,115.56,115.21,114.04,113.64,113.22,110.17,101.08,62.27,50.96。
实施例5
化合物8d的制备:
将实施例1得到的中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮0.317g和2,4-二氯苯氧乙炔0.201g溶解在体积比3:1的叔丁醇∶水的20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在温度80℃下反应过夜,待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.477g化合物8d,名称为5-((4-((2,4-二氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-((2,4-二氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物8d)的核磁数据:
1hnmr(400mhz,cdcl3)δ8.03(s,1h),7.86(s,1h),7.75(s,1h),7.59–7.50(m,5h),7.48–7.41(m,1h),7.32(d,j=2.4hz,1h),7.14(dd,j=8.8,2.5hz,1h),7.00(d,j=8.8hz,1h),6.03(s,1h),5.81(s,2h),5.30(s,2h).
13cnmr(101mhz,cdcl3)δ161.13,157.53,155.69,152.48,148.16,143.47,142.31,131.06,130.29,129.53,128.99,128.47,127.87,127.65,125.78,124.70,123.58,115.35,115.14,113.98,110.98,101.09,63.68,50.95。
实施例6
化合物8e的制备:
将实施例1得到的中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮0.317g和3,5-二氯苯氧乙炔0.201g溶解在体积比3:1的叔丁醇∶水的20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在温度80℃下反应过夜,待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.450g化合物8e,5-((4-((3,5-二氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-((3,5-二氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物8e)的核磁数据:
1hnmr(400mhz,cdcl3)δ8.04(s,1h),7.86(s,1h),7.70(s,1h),7.57–7.51(m,5h),7.48–7.41(m,1h),6.97(t,j=1.8hz,1h),6.87(d,j=1.7hz,2h),6.07(s,1h),5.82(s,2h),5.20(s,2h).
13cnmr(101mhz,cdcl3)δ160.04,157.32,156.52,152.79,148.11,143.48,131.06,129.54,129.41,128.99,128.49,127.66,124.73,123.47,121.98,115.20,114.06,113.77,109.69,101.10,62.44,50.98。
实施例7
化合物8f的制备:
将实施例1得到的中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮0.317g和3,4-二氯苯氧乙炔0.201g溶解在体积比3:1的叔丁醇∶水的20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在温度80℃下反应过夜,待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.463g化合物8f,名称为5-((4-((3,4-二氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-((3,4-二氯苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物8f)的核磁数据:
1hnmr(400mhz,cdcl3)δ8.04(s,1h),7.86(s,1h),7.71(s,1h),7.59–7.52(m,5h),7.49–7.41(m,1h),7.30(d,j=8.9hz,1h),7.07(d,j=2.9hz,1h),6.82(dd,j=9.0,2.9hz,1h),6.06(s,1h),5.81(s,2h),5.20(s,2h).
13cnmr(101mhz,cdcl3)δ160.05,157.54,157.10,156.31,152.08,148.13,143.49,130.95,129.53,128.48,127.65,124.72,123.47,122.49,117.05,115.19,114.77,114.05,113.60,110.15,101.09,62.46,50.98。
实施例8
化合物8g的制备:
将0.317g中间体7和0.150g4-氟苯氧乙炔溶解在体积比3:1的叔丁醇∶水的20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在80℃下反应过夜。待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.415g化合物8g,5-((4-((4-氟苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-((4-氟苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物)的核磁数据:
1hnmr(400mhz,cdcl3)δ8.04(s,1h),7.86(s,1h),7.71(s,1h),7.59–7.50(m,6h),7.48–7.41(m,1h),6.99–6.85(m,4h),6.04(s,1h),5.80(s,2h),5.19(s,2h).
13cnmr(101mhz,cdcl3)δ160.06,157.50,156.54,154.20,152.05,148.24,143.45,131.02,129.53,128.93,128.46,127.65,124.67,123.40,122.48,116.21,115.98,115.20,113.95,113.63,101.03,62.71,50.90.
实施例9
化合物8h的制备:
将实施例1得到的中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮0.317g和2-氟苯氧乙炔0.150g溶解在体积比3:1的叔丁醇∶水的20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在温度80℃下反应过夜,待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.406g化合物8h,名称为5-((4-((2-氟苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-((2-氟苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物8h)的核磁数据:
1hnmr(400mhz,cdcl3)δ8.03(s,1h),7.85(s,1h),7.71(s,1h),7.61–7.49(m,5h),7.47–7.40(m,1h),7.12–6.96(m,3h),6.96–6.85(m,1h),6.01(s,1h),5.80(s,2h),5.30(s,2h).
13cnmr(101mhz,cdcl3)δ160.03,157.50,154.29,152.06,148.21,146.00,143.42,132.45,131.05,130.56,129.52,128.97,128.45,127.66,124.58,122.41,116.46,115.19,113.94,113.66,101.03,63.65,50.94。
实施例10
化合物8i的制备:
将实施例1得到的中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮0.317g和3,5-二氟苯氧乙炔0.168g溶解在体积比3:1的叔丁醇∶水的20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在温度80℃下反应过夜,待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.438g化合物8i,名称为5-((4-((3,5-二氟苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-((3,5-二氟苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物8i)的核磁数据:
1hnmr(400mhz,cdcl3)δ8.05(s,1h),7.86(s,1h),7.72(s,1h),7.59–7.52(m,5h),7.47–7.43(m,1h),6.50(d,j=8.8hz,2h),6.43(t,j=8.9hz,1h),6.06(s,1h),5.82(s,2h),5.19(s,2h).
13cnmr(101mhz,cdcl3)δ160.04,157.54,155.60,152.09,148.12,143.48,131.06,129.54,128.99,128.49,127.66,124.74,123.02,122.53,115.20,114.05,113.58,109.19,101.09,62.49,50.98。
实施例11
化合物8j的制备:
将实施例1得到的中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮0.317g和4-溴苯氧乙炔0.211g溶解在体积比3:1的叔丁醇∶水的20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在温度80℃下反应过夜,待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.479g化合物8j,名称为5-((4-((4-溴苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-((4-溴苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物8j)的核磁数据:
1hnmr(400mhz,cdcl3)δ8.04(s,1h),7.86(s,1h),7.71(s,1h),7.59–7.51(m,5h),7.48–7.41(m,1h),7.35(d,j=8.9hz,2h),6.84(d,j=8.9hz,2h),6.05(s,1h),5.80(s,2h),5.20(s,2h).
13cnmr(101mhz,cdcl3)δ160.06,157.52,157.21,153.19,152.07,148.17,143.47,132.54,129.54,128.48,127.65,124.69,123.41,122.49,116.75,115.20,114.03,113.85,113.62,101.07,62.25,50.95。
实施例12
化合物8k的制备:
将实施例1得到的中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮0.317g和苯氧乙炔0.132g溶解在体积比3:1的叔丁醇∶水的20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在温度80℃下反应过夜,待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.407g化合物8k,名称为5-((4-(苯氧基甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-(苯氧基甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物8k)的核磁数据:
1hnmr(400mhz,cdcl3)δ8.05(s,1h),7.86(s,1h),7.72(s,1h),7.61–7.50(m,5h),7.48–7.41(m,1h),7.28(d,j=8.0hz,2h),6.96(d,j=8.0hz,3h),6.05(s,1h),5.80(s,2h),5.24(s,2h).
13cnmr(101mhz,cdcl3)δ160.07,158.12,157.51,152.08,148.24,143.42,130.57,129.72,129.53,128.46,127.67,124.68,123.32,122.52,121.59,115.23,114.91,114.00,113.67,101.03,62.09,50.90。
实施例13
化合物8l的制备:
将实施例1得到的中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮0.317g和2-甲基苯氧乙炔0.146g溶解在体积比3:1的叔丁醇∶水的20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在温度80℃下反应过夜,待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.420g化合物8l,名称为5-((4-((2-甲基苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-((2-甲基苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物8l)的核磁数据:
1hnmr(400mhz,cdcl3)δ8.05(s,1h),7.86(s,1h),7.68(s,1h),7.61–7.50(m,5h),7.48–7.41(m,1h),7.08–7.17(m,2h),6.96–6.83(m,2h),6.04(s,1h),5.81(s,2h),5.25(s,2h),2.19(s,3h).
13cnmr(101mhz,cdcl3)δ160.09,157.51,156.28,152.08,148.31,143.39,131.02,130.57,129.53,128.46,127.67,127.01,124.68,122.52,121.31,115.19,113.89,113.69,111.70,101.07,62.41,50.92,29.86。
实施例14
化合物8m的制备:
将实施例1得到的中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮0.317g和4-甲基苯氧乙炔0.146g溶解在体积比3:1的叔丁醇∶水的20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在温度80℃下反应过夜,待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.411g化合物8m,名称为5-((4-((4-甲基苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-((4-甲基苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物8m)的核磁数据:
1hnmr(400mhz,cdcl3)δ8.04(s,1h),7.86(s,1h),7.72(s,1h),7.60–7.51(m,5h),7.48–7.41(m,1h),7.06(d,j=8.1hz,2h),6.85(d,j=8.4hz,2h),6.05(s,1h),5.79(s,2h),5.20(s,2h),2.27(s,3h).
13cnmr(101mhz,cdcl3)δ160.07,157.50,156.00,152.08,148.23,143.45,143.37,130.89,130.57,130.15,129.54,128.46,127.67,124.67,122.52,115.28,115.23,114.81,114.06,113.99,113.68,101.05,62.26,50.98,29.85。
实施例15
化合物8n的制备:
将实施例1得到的中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮0.317g和2-甲氧基苯氧乙炔0.162g溶解在体积比3:1的叔丁醇∶水的20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在温度80℃下反应过夜,待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.429g化合物8n,名称为5-((4-((2-甲氧基苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-((2-甲氧基苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物8n)的核磁数据:
1hnmr(600mhz,cdcl3)δ8.04(s,1h),7.86(s,1h),7.71(s,1h),7.59–7.51(m,5h),7.48–7.41(m,1h),6.99(d,j=8.0hz,1h),6.95–6.90(m,1h),6.89–6.81(m,2h),6.05(s,1h),5.78(s,2h),5.32(s,2h).
13cnmr(101mhz,cdcl3)δ160.06,157.52,156.69,152.11,148.21,143.39,145.97,130.57,130.18,129.54,128.45,127.68,124.67,123.44,122.39,121.02,115.30,114.94,114.09,113.70,112.08,101.02,63.48,55.96,50.90。
实施例16
化合物8o的制备:
将实施例1得到的中间体7为5-叠氮甲基-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮0.317g和2-甲氧基苯氧乙炔0.162g溶解在体积比3:1的叔丁醇∶水的20ml混合溶剂中,再加入0.44g五水硫酸铜和0.06g铜粉,在温度80℃下反应过夜,待原料全部消失后,冷却至室温,加入水蒸馏水,用二氯甲烷萃取,有机相干燥,浓缩,将残渣采用柱层析梯度洗脱,洗脱剂为体积比2∶1的石油醚∶乙酸乙酯,最终得到0.415g化合物8o,名称为5-((4-((4-甲氧基苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮。
5-((4-((4-甲氧基苯氧基)甲基)-1h-1,2,3-三氮唑-1-基)甲基)-3-苯基-7h-呋喃[3,2-g]苯并吡喃-7-酮(化合物8o)的核磁数据:
1hnmr(400mhz,cdcl3)δ8.04(s,1h),7.86(s,1h),7.71(s,1h),7.60–7.50(m,5h),7.48–7.41(m,1h),6.93(d,j=9.1hz,2h),6.84(d,j=9.1hz,2h),6.03(s,1h),5.79(s,2h),5.19(s,2h),3.77(s,3h).13cnmr(101mhz,cdcl3)δ160.07,157.50,154.47,152.19,151.82,148.25,143.46,130.57,129.54,128.46,127.67,124.67,122.51,116.29,116.08,115.20,114.84,114.00,113.67,101.06,62.88,55.79,50.98。
实施例17
本发明所述的如结构式所示的含苯基三氮唑的补骨脂素衍生物(8a-8o)对b16细胞中黑素合成及酪氨酸酶活性的作用测定:
筛选模型:小鼠b16黑素瘤细胞
细胞来源:中科院细胞库提供;
(1)细胞毒性试验(mtt法):
将正处于对数生长期的黑素细胞接种于96孔板中,每孔细胞数为5×103/孔,每个浓度设8复孔,24h后(细胞贴壁后)、加入不同浓度的样品,48h后处理细胞、每孔加入10μl的mtt(浓度为5mg/ml)后在温度37℃培养箱中培养4h,取出96孔板后每孔加入150μl的dmso并震荡10min,等到结晶全部溶解后用酶标仪在570nm处测定吸光度(a570),未加药的孔作为对照组、细胞活性按100%计算,而加药组实验数据最少重复3遍;
(2)蛋白定量用bradford法测定:
完全溶解蛋白标准品(5mg/mlbsa),取10μl稀释至100μl,使终浓度为0.5mg/ml。蛋白样品在什么溶液中,标准品也宜用什么溶液稀释,但是为了简便起见,也可以用0.9%nacl或pbs稀释标准品;将稀释后标准品(0.5mg/mlbsa)按0,1,2,4,8,12,16,20μl分别加到96孔板中,加标准品稀释液将所有标准品补足到20μl;加适当体积样品到96孔板的样品孔中,加标准品稀释液补足到20μl;各孔加入200μlbradford染色液,用加样枪轻轻吹打混匀(注意不要弄出气泡影响读数)室温放置3-5min;用酶标仪测定a595;根据标准曲线计算出样品中的蛋白浓度;
(3)黑素含量用碱消化法进行测定:
将处于对数生长期的黑素细胞接种于6cm培养皿中、浓度为5×105个/ml,各孔加5ml细胞悬浮液;接种12h后、待细胞完全贴壁后用药,用药浓度分别为1、5、10、20、50和100μg/ml;在72h后收集细胞;在不破碎细胞的情况下加入200μl的1mnaoh溶液,置温度80℃水浴中2h后,测定波长470nm处的吸光度(a470值),未用药组作为对照组与用药组进行对比;
(4)酪氨酸酶活性用l-dopa染色法进行测定:
正处于对数生长期的黑素细胞接种于12孔板中、浓度为2×105个/ml,各孔加1ml细胞悬浮液;接种12h后、待细胞完全贴壁后用药,用药浓度分别为1、5、10、20、50和100μg/ml;在48h后收集细胞;处理完细胞后每个ep管中加tris-0.1%triton-100溶液200μl,放入-20℃冰箱中30min-1h进行裂解;用bradford法进行蛋白含量测定;将样品蛋白含量调到统一浓度;各反应体系加入100μl的0.1%l-dopa后放到温度37℃培养箱中,培养2h后在475nm处测定吸光度(a475)。未用药的组作为对照组与用药组进行对比。
结果:化合物8a、8j、8m-8o共5个补骨脂素胺类衍生物均可用于临床上制备治疗白癜风的药物的用途。
- 还没有人留言评论。精彩留言会获得点赞!
- 用于生产化学品及其衍生物的组合物和方法与制造工艺
- 7‑OH‑8‑[(1‑胺基‑1‑苯基衍生物)‑次甲基]‑黄酮化合物及其制备方法及其应用与制造工艺
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 含D型非天然氨基酸的抗菌肽类似物及其合成与应用的制造方法与工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 一种白杨素氨基酸衍生物的制备方法与制造工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 吡唑衍生物及其作为大麻素受体介体的用途的制造方法与工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 水生态处理中淤泥及衍生物的再利用处理系统的制造方法与工艺
- Schiglautone A衍生物的组合物用于制备抗缺氧药物的制作方法
- Schiglautone A衍生物的组合物用于制备抗红细胞低下性贫血药物的制作方法
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 水生态处理中淤泥及衍生物的再利用处理系统的制造方法与工艺
- 一种新型罗汉果醇衍生物单体的制作方法
- Schiglautone A衍生物的组合物用于制备抗鼻炎药物的制作方法
- Schiglautone A衍生物的组合物用于制备防治肾纤维化药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗心衰药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Schiglautone A 衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备防治胰腺纤维化药物的制作方法