一种基于ddPCR定量检测饲料中转基因玉米IE034的方法、引物和探针及应用与流程
本发明属于分子生物学及转基因检测检测技术领域,具体涉及饲料中转基因玉米ie034的微滴式数字pcr定量检测方法。
背景技术:
2016年,全球转基因作物的种植面积从1996年的170万公顷增加到1.851亿公顷,其中转基因玉米的种植面积为6060万公顷,占全球玉米总种植面积的64%,应用率为26%。截止到2016年,共有40个国家/地区的监管机构批准转基因作物用作粮食和/或饲料以及释放到环境中,在获准的3768项监管审批中有1238项涉及饲料用途,其中包括直接用途或加工用途,用于饲料的转基因作物占比约为33%。玉米是重要的饲料原料,随着饲料加工及工业酒精对玉米需求的增长,转基因玉米在转基因作物种植中所占比重还会逐年增加。我国是转基因玉米进口大国,2015年进口量为330万吨玉米,目前正在加快转基因作物种植的审批,以提高玉米生产率并且减少对数量不断增长的进口转基因玉米的依赖。
转基因作物商业化种植给全球带来了巨大的社会和经济效益,同时其潜在的安全问题也引起了人们的极大关注。目前,已有60多个国家制定了对含有转基因成分或由转基因作物加工而成的产品进行标识的制度,而且随着消费者对转基因产品关注度的日益提高,更加强调了对转基因成分进行定量检测的重要性,如何准确、精确地确定转基因作物及其产品中的外源基因,已经成为现今转基因产品检测技术研究的一个关键点,也是转基因生物安全评价的重要参数。
目前,通常采用实时荧光定量pcr(qpcr)对转基因玉米ie034中的转基因成分进行定量检测。经检索,现有技术已经公布了相关的技术方案,中国专利号201410457457.5,公开日为2014.12.10的申请案公开了转基因玉米ie034的转化体特异性定量pcr检测引物和探针及方法,其设计了上游引物序列、下游引物序列及荧光探针序列进行pcr扩增,虽然该实时荧光定量pcr的方法能够对转基因玉米ie034及其衍生品种进行特异性定量检测,然而依然存在一定的缺陷:在分析外源基因拷贝数时,实时荧光定量pcr必须依赖于标准曲线和已知拷贝数的基因,只是一种相对定量方法,且标准曲线的质量易受到dna纯度、引物和探针的浓度、反应抑制因子等诸多因素影响,使得检测灵敏度及精确度都达不到准确定量的要求,可能导致过低或过高估计转基因的含量。而且,由于外源基因通常是以低拷贝数(1~2个)整合到受体基因组中,在检测复杂物质中的转基因成分时,qpcr容易受到背景dna的干扰。而近年来兴起的微滴式数字pcr(ddpcr)是一种新的绝对定量技术,通过极度稀释实现理论上的单分子扩增,使得稀有的目标核酸序列与大量的背景dna分开,提高其相对含量,进而提高了检测的灵敏度及重复性,目前该技术在转基因玉米ie034的检测领域未得到应用。
因此,基于现有技术的缺陷,亟需提供一种对饲料中转基因玉米ie034精准定量检测的方法。
技术实现要素:
1.解决的技术问题:
基于现有pcr技术对于饲料中转基因玉米ie034的检测灵敏度及精确度都达不到准确定量的要求,本发明提供一种对饲料中转基因玉米ie034精准定量检测的方法。
2.技术方案:
本发明的目的是通过以下技术方案实现的:
本发明提供了一种基于ddpcr定量检测饲料中转基因玉米ie034的引物和探针,包括:
外源基因cry1ie正向引物:5′-agagctgggtgcgctacaac-3′
外源基因cry1ie反向引物:5′-acaccagggtgtcgtagctg-3′
外源基因cry1ie探针:5′-fam-ttccgcaaggacatgaccctgatggt-tamar-3′
内参基因rubisco正向引物:5′-atgtagcttatccattagac-3′
内参基因rubisco反向引物:5′-ttgaaaccaaatacgttacc-3′
内参基因rubisco探针:5′-fam-atttgaagagggttctgttactaacat-tamar-3′
作为本发明更进一步的改进,使用引物和探针进行pcr扩增。
作为本发明更进一步的改进,本发明还提供了一种基于ddpcr定量检测饲料中转基因玉米ie034的方法,包括以下步骤:
1)、提取饲料样品的基因组dna;
2)、ddpcr扩增:以步骤1)所述的基因组dna为模板,利用所述的引物和探针,分别根据外源基因cry1ie和内参基因rubisco设定相应的数字pcr扩增反应体系和反应程序,进行扩增反应,得到pcr扩增产物;
3)、对步骤2)得到的pcr扩增产物进行检测与分析。
作为本发明更进一步的改进,扩增基因是外源基因cry1ie时,将基因组dna浓度稀释至10~100ng/μl;扩增基因是玉米内参基因rubisco时,将基因组dna浓度稀释至1~0.1ng/μl。
作为本发明更进一步的改进,所述步骤2)中引物和探针组合中引物浓度为10μmol/l,探针浓度为5μmol/l。
作为本发明更进一步的改进,所述的外源基因cry1ie的数字pcr扩增反应体系包括:2xddpcrsupermixforprobes10μl,上下游引物各0.4μl,浓度为10μmol/l,探针0.6μl,浓度为5μmol/l,dna模板2.5μl,无菌去离子水补足至20μl;所述的内参基因rubisco的数字pcr扩增反应体系包括:2xddpcrsupermixforprobes13.5μl,上下游引物各0.3μl,浓度为10μmol/l,探针0.2μl,浓度为5μmol/l,dna模板3μl,无菌去离子水补足至20μl。
作为本发明更进一步的改进,外源基因cry1ie的数字pcr扩增反应程序为:95℃预变性120s,95℃变性10s,59℃退火30s,72℃延伸30s,总共47个循环;扩增内参基因rubisco的数字pcr扩增反应程序为:95℃预变性600s,95℃变性15s,54℃退火30s,72℃延伸30s,共45个循环。
作为本发明更进一步的改进,所述的步骤3)包括以下步骤:
a)将所得的pcr产物置于微滴分析仪内,并在quantsoft软件上设定检测模式为abs,检测fam的荧光信号;
b)微滴分析仪对每个微滴逐个进行荧光检测和分析;
c)由quantsoft计算各饲料样品中外源基因和内参基因的拷贝数浓度。
作为本发明更进一步的改进,所述方法应用于饲料中转基因玉米ie034的定量检测。
3.有益效果:
与现有技术相比,本发明的有益效果在于:
(1)本发明的基于ddpcr定量检测饲料中转基因玉米ie034的引物和探针,能够实现对饲料中外源基因和玉米内参基因的特异性扩增,与现有技术中的转基因玉米ie034的pcr扩增相比,大幅度提高了检测方法的准确度和灵敏度。
(2)本发明的基于ddpcr定量检测饲料中转基因玉米ie034的方法,针对于现有技术中采用荧光定量pcr检测饲料中转基因玉米ie034时,容易受背景dna中抑制剂的干扰,以及不同样品和试验过程中pcr扩增效率变化的影响,导致灵敏度低、准确度较差的缺陷,实现了饲料中转基因玉米ie034基因组dna的ddpcr扩增,该方法条件下pcr扩增产物满足ddpcr的检测条件,最终实现了饲料中转基因玉米ie034外源基因的绝对定量检测,检测过程中不依赖标准曲线、不受背景dna中抑制剂的干扰及不同样品和试验过程中pcr扩增效率变化的影响,具有更高的灵敏度和准确度。
(3)本发明的基于ddpcr定量检测饲料中转基因玉米ie034的方法,根据外源基因和内参基因rubisco的特点,对基因组dna进行稀释,在扩增基因是外源基因cry1ie时,基因组dna浓度范围为10~100ng/μl;扩增基因是玉米内参基因rubisco时,基因组dna浓度范围1~0.1ng/μl,在上述基因组dna浓度范围内可达到ddpcr的扩增倍数,满足检测条件,该方法对基因组dna处理方法简单,利于推广。
(4)本发明的基于ddpcr定量检测饲料中转基因玉米ie034的方法,分别根据外源基因cry1ie和内参基因rubisco的引物和探针的特点设定ddpcr扩增反应体系和反应程序,保证采用的ddpcr扩增反应体系和反应程序适用于本发明设计的引物和探针,在合适的基因组dna模板条件下实现ddpcr的扩增反应,具有针对性强的特点,进一步提高最终检测的准确度和灵敏度。
(5)本发明的基于ddpcr定量检测饲料中转基因玉米ie034的方法,该方法可以实现转基因玉米ie034及其产品中外源基因的定量检测,为转基因产品的分析与检测提供一定的指导价值。
附图说明
图1为实验组饲料样品基因组dna浓度为100ng/μl时外源基因的ddpcr扩增结果;
图2为实验组饲料样品基因组dna浓度为10ng/μl时外源基因的ddpcr扩增结果;
图3为实验组饲料样品基因组dna浓度为1ng/μl时外源基因的ddpcr扩增结果;
图4为实验组饲料样品基因组dna浓度为10ng/μl时内参基因的ddpcr扩增结果;
图5为实验组饲料样品基因组dna浓度为1ng/μl时内参基因的ddpcr扩增结果;
图6为实验组饲料样品基因组dna浓度为0.1ng/μl时内参基因的ddpcr扩增结果;
图7为对照组饲料样品基因组dna浓度为100ng/μl时外源基因的ddpcr扩增结果;
图8为对照组饲料样品基因组dna浓度为10ng/μl时外源基因的ddpcr扩增结果;
图9为对照组饲料样品基因组dna浓度为1ng/μl时外源基因的ddpcr扩增结果;
图10为对照组饲料样品基因组dna浓度为10ng/μl时内参基因的ddpcr扩增结果;
图11为对照组饲料样品基因组dna浓度为1ng/μl时内参基因的ddpcr扩增结果;
图12为对照组饲料样品基因组dna浓度为0.1ng/μl时内参基因的ddpcr扩增结果。
具体实施方式
下面将对本发明实施例作进一步地详细描述。
实施例1
1、实验用饲料配比:
实验用饲料分为实验组和对照组两种,实验组饲料玉米成分为转基因玉米ie034,对照组饲料玉米成分为近等基因系的非转基因玉米综31,实验组和对照组饲料配比方案如表1所示。
表1实验组和对照组饲料配比方案
其中,鱼粉厂家为山东天成生物科技有限公司生产的秘鲁鱼粉;酪蛋白厂家为sigma;明胶的厂家为sigma;鱼肝油的厂家为挪威axellusas,orkla;复合维生素矿物质的厂家为新乡八牧科技有限公司;加丽素红10%的厂家为帝斯曼营养产品法国有限公司。
本实验所用到的转基因玉米ie034及其亲本非转基因玉米综31均由中国农业科学院作物研究所提供。
2、饲料基因组dna提取
采用动物源性植物饲料基因组dna提取试剂盒dp323,该试剂盒购买自天根生化科技有限公司,利用dna提取试剂盒dp323分别提取实验组和对照组饲料基因组dna:
1)将配制好的饲料研磨粉碎,取50mg粉碎后的饲料样品加入320μl缓冲液ga,振荡至彻底悬浮;
2)加入20μl蛋白酶k溶液,混匀,在65℃放置20~30min,其间混匀样品2~3次;
3)加入340μl缓冲液gb,充分颠倒混匀,65℃放置10min,其间混匀样品2~3次;
4)以12000rpm的转速离心2min,取440μl上清液至新管中;
5)向上清液中加入220μl无水乙醇,充分颠倒混匀6~8次;
6)将上一步所得溶液和絮状沉淀全部加入一个吸附柱cb3中(吸附柱放入收集管中),12000rpm离心2min,倒掉废液,将吸附柱cb3放回收集管中;
7)向吸附柱cb3中加入500μl缓冲液gd,12000rpm离心30s,倒掉废液,将吸附柱cb3放回收集管中;
8)向吸附柱cb3中加入600μl漂洗液pw,12000rpm离心30s,倒掉废液。将吸附柱cb3放入收集管中;
9)向吸附柱cb3中加入500μl缓冲液gd,12000rpm离心30s,倒掉废液,将吸附柱cb3放回收集管中;
10)将吸附柱cb3放回收集管中,12000rpm离心2min,倒掉废液,将吸附柱cb3置于室温放置数分钟,以彻底晾干吸附材料中残余的漂洗液;
11)将吸附柱cb3转入一个干净的离心管中,向吸附膜的中间部位悬空滴加50μl洗脱缓冲液te,室温放置2~5min,12000rpm离心2min。
3、引物和探针
用以下引物和探针组合进行数字pcr扩增,引物和探针均由宝生物工程(大连)有限公司合成,其中外源基因cry1ie的探针5′标记发光基团fam,3′标记淬灭基团tamar,内参基因rubisco探针5′标记发光基团fam,3′标记淬灭基团tamar:
外源基因cry1ie正向引物:5′-agagctgggtgcgctacaac-3′
外源基因cry1ie反向引物:5′-acaccagggtgtcgtagctg-3′
外源基因cry1ie探针:5′-fam-ttccgcaaggacatgaccctgatggt-tamar-3′
内参基因rubisco正向引物:5′-atgtagcttatccattagac-3′
内参基因rubisco反向引物:5′-ttgaaaccaaatacgttacc-3′
内参基因rubisco探针:5′-fam-atttgaagagggttctgttactaacat-tamar-3′
4、ddpcr扩增
采用紫外分光光度计定量分析提取的实验组和对照组饲料样品基因组dna的浓度和纯度,结果表明,实验组和对照组饲料样品基因组dna的od260nm/od280nm比值均在1.7至1.9之间,实验组和对照组饲料样品基因组dna的浓度均约为100ng/μl。
ddpcr扩增前对实验组和对照组饲料样品基因组dna进行稀释,扩增外源基因cry1ie时分别将样品基因组dna稀释成100ng/μl、10ng/μl和1ng/μl三个浓度梯度;扩增玉米内参基因rubisco时将样品基因组dna稀释成10ng/μl、1ng/μl和0.1ng/μl三个浓度梯度。
将上述的引物和探针用te缓冲溶液进行溶解稀释,引物稀释后的浓度为10μmol/l,探针稀释后的浓度为5μmol/l。
以稀释后的基因组dna为模板,利用稀释后的引物和探针组合,配制相应的ddpcr扩增反应体系,设定反应程序,每组样品分别设置3个平行进行ddpcr扩增反应,得到pcr扩增产物。稀释后的基因组dna模板浓度与扩增基因的对应关系如表2所示。
表2基因组dna模板浓度与扩增基因对应关系
外源基因cry1ie的数字pcr扩增反应体系如表3所示。
表3外源基因cry1ie的数字pcr扩增反应体系
内参基因rubisco的数字pcr扩增反应体系如表4所示。
表4内参基因rubisco的数字pcr扩增反应体系
将配制好的样品反应体系加入微滴发生卡的样品孔中,注意加样过程中避免产生气泡,再将70μl的微滴发生油加入到反应油孔内,将密封垫盖在发生卡底座上,置于微滴发生器中生成油包水的微滴,从微滴发生卡上层反应孔内吸取40μl微滴,转移至96孔pcr反应板的一个反应孔内,注意避免微滴的破坏和聚集,封膜仪180℃热封,使用热封铝膜封口后置于pcr仪上设定反应程序进行pcr扩增。
外源基因的pcr扩增反应程序为:95℃预变性120s,95℃变性10s,59℃退火30s,72℃延伸30s,总共47个循环;内参基因的pcr扩增反应程序为:95℃预变性600s,95℃变性15s,54℃退火30s,72℃延伸30s,共45个循环。
5、ddpcr结果读取与分析
ddpcr反应结束后,将扩增后的pcr反应板置于微滴分析仪(qx200,bio-rad,usa)内,打开quantsoft软件,点击flushsystem键冲洗系统,然后点击setup键输入样品信息,检测模式设定为abs,检测fam的荧光信号,输入完成后点击run,微滴分析仪会自动对每个微滴进行荧光检测和分析,quantsoft软件根据检测到的阳性微滴和阴性微滴的比例,按照泊松分布原理计算获得样品中外源基因和内参基因的拷贝数浓度。
微滴生成是微滴式数字pcr的关键步骤,只有微滴数大于11000时,dna分子的分布才能符合泊松分布的统计学原理,系统才可以对阳性微滴和阴性微滴进行计数。
图1为实验组饲料样品基因组dna浓度为100ng/μl时,外源基因cry1ie的扩增图,图中上半部分的黑色信号点代表发生pcr扩增的微滴,系统判读为阳性信号,阳性微滴数为2883;下半部分的黑色信号点代表未发生pcr扩增的微滴,系统判读为阴性信号,阴性微滴数为13171。
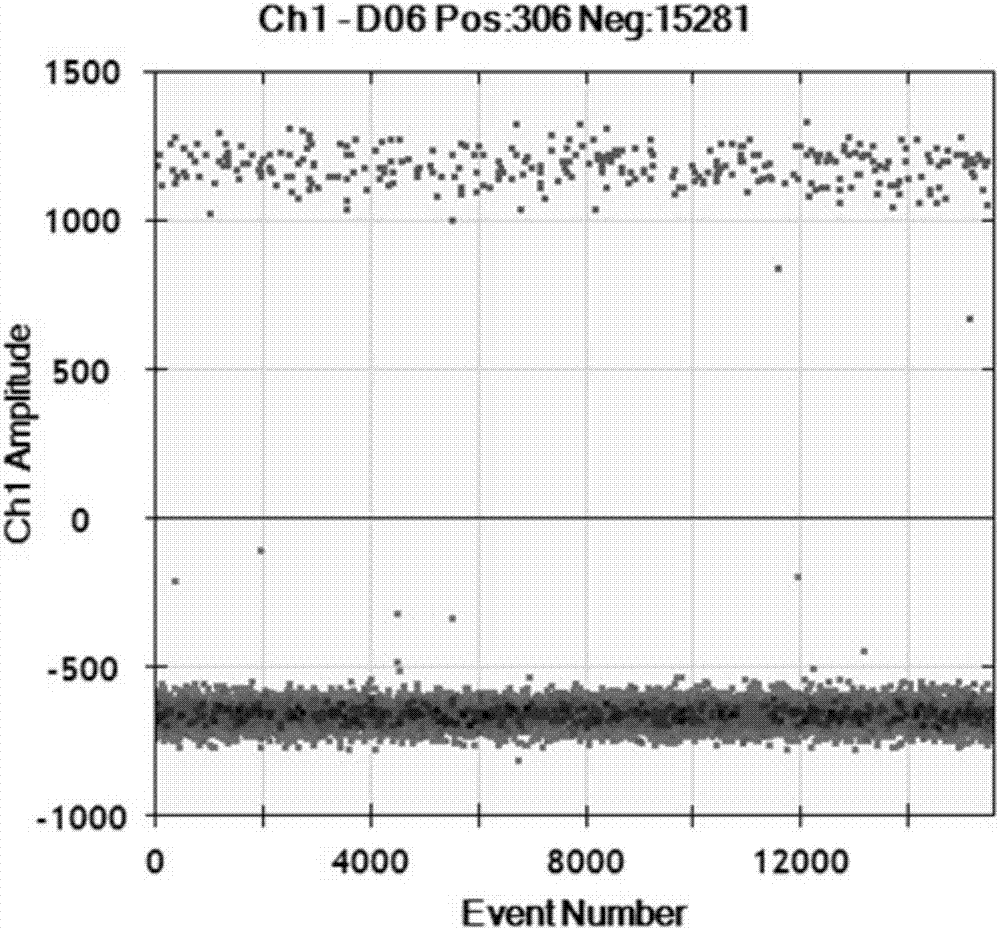
图2为实验组饲料样品基因组dna浓度为10ng/μl时,外源基因cry1ie的扩增图,图中上半部分的黑色信号点代表发生pcr扩增的微滴,系统判读为阳性信号,阳性微滴数为306;下半部分的黑色信号点代表未发生pcr扩增的微滴,系统判读为阴性信号,阴性微滴数为15281。
图3为实验组饲料样品基因组dna浓度为1ng/μl时,外源基因cry1ie的扩增图,系统未检测到阳性信号,图中显示阳性微滴数为0;黑色信号点代表未发生pcr扩增的微滴,系统判读为阴性信号,阴性微滴数为15938。
图4为实验组饲料样品基因组dna浓度为10ng/μl时,内参基因rubisco的扩增图,系统未检测到阳性信号,图中显示阳性微滴数为0;黑色信号点代表未发生pcr扩增的微滴,系统判读为阴性信号,阴性微滴数为15656。
图5为实验组饲料样品基因组dna浓度为1ng/μl时,内参基因rubisco的扩增图,图中上半部分的黑色信号点代表发生pcr扩增的微滴,系统判读为阳性信号,阳性微滴数为15454;下半部分的黑色信号点代表未发生pcr扩增的微滴,系统判读为阴性信号,阴性微滴数为3344。
图6为实验组饲料样品基因组dna浓度为0.1ng/μl时,内参基因rubisco的扩增图,图中上半部分的黑色信号点代表发生pcr扩增的微滴,系统判读为阳性信号,阳性微滴数为2158;下半部分的黑色信号点代表未发生pcr扩增的微滴,系统判读为阴性信号,阴性微滴数为12620。
图7为对照组饲料样品基因组dna浓度为100ng/μl时,外源基因cry1ie的扩增图,系统未检测到阳性信号,图中显示阳性微滴数为0;黑色信号点代表未发生pcr扩增的微滴,系统判读为阴性信号,阴性微滴数为15110。
图8为对照组饲料样品基因组dna浓度为10ng/μl时,外源基因cry1ie的扩增图,系统未检测到阳性信号,图中显示阳性微滴数为0;黑色信号点代表未发生pcr扩增的微滴,系统判读为阴性信号,阴性微滴数为15034。
图9为对照组饲料样品基因组dna浓度为1ng/μl时,外源基因cry1ie的扩增图,系统未检测到阳性信号,图中显示阳性微滴数为0;黑色信号点代表未发生pcr扩增的微滴,系统判读为阴性信号,阴性微滴数为14442。
图10为对照组饲料样品基因组dna浓度为10ng/μl时,内参基因rubisco的扩增图,系统未检测到阳性信号,图中显示阳性微滴数为0;黑色信号点代表未发生pcr扩增的微滴,系统判读为阴性信号,阴性微滴数为13600。
图11为对照组饲料样品基因组dna浓度为1ng/μl时,内参基因rubisco的扩增图,图中上半部分的黑色信号点代表发生pcr扩增的微滴,系统判读为阳性信号,阳性微滴数为14016;下半部分的黑色信号点代表未发生pcr扩增的微滴,系统判读为阴性信号,阴性微滴数为2087。
图12为对照组饲料样品基因组dna浓度为0.1ng/μl时,内参基因rubisco的扩增图,图中上半部分的黑色信号点代表发生pcr扩增的微滴,系统判读为阳性信号,阳性微滴数为3439;下半部分的黑色信号点代表未发生pcr扩增的微滴,系统判读为阴性信号,阴性微滴数为14499。
将本实施例ddpcr反应中形成的总微滴数统计,统计结果如表5所示。
表5实验组和对照组饲料样品ddpcr扩增形成的总微滴数
由表5可知,生成的微滴数处于12856~18930之间,均大于11000,满足微滴式数字pcr微滴的分析要求。
根据quantsoft软件读取实验组和对照组饲料样品中外源基因和内参基因的拷贝数浓度如表6所示,其中,nocall表示未检测出。
表6实验组和对照组饲料样品中外源基因和内参基因的拷贝数
由表6可知,基因组dna浓度为100ng/μl时,实验组饲料样品中外源基因cry1ie的拷贝数浓度平均值为240.33copies/μl;基因组dna浓度为10ng/μl时,外源基因cry1ie的拷贝数浓度平均值为23.63copies/μl;基因组dna浓度为1ng/μl时,实验组饲料样品中未检测到外源基因;结果表明对饲料样品中的外源基因cry1ie进行ddpcr定量检测时,模板dna浓度应为10~100ng/μl。对照组饲料样品中均未检测到外源基因,说明数字pcr的结果与预期相符。
基因组dna浓度为10ng/μl时,实验组和对照组饲料样品中均未检测到玉米内参基因rubisco;基因组dna浓度为1ng/μl时,实验组和对照组饲料样品中内参基因的拷贝数浓度平均值分别为2099.67copies/μl和2221.67copies/μl;基因组dna浓度为0.1ng/μl时,内参基因的拷贝数浓度平均值分别为216.67copies/μl和219.33copies/μl;结果说明对饲料样品中的玉米内参基因rubisco进行ddpcr定量检测时,模板dna的浓度应为0.1~1ng/μl。
本发明的结果表明,ddpcr能够精准定量检测饲料样品中的外源基因和内参基因,具有良好的重复性和稳定性。
以上示意性的对本发明及其实施方式进行了描述,该描述没有限制性,附图中所示的也只是本发明的实施方式之一,实际的流程并不局限于此。所以,如果本领域的普通技术人员受其启示,在不脱离本发明创造宗旨的情况下,不经创造性的设计出与该技术方案相似的结构方式及实施例,均应属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 基于环介导等温扩增消光技术的转基因玉米59122检测方法与流程
- 鉴定转基因植物或者转基因玉米双抗12‑5外源基因插入染色体位置的方法及其应用与流程
- CYP78A基因在增加玉米株高和增强植株长势中的应用的制作方法与工艺
- 转基因玉米次生代谢物的HPLC‑MS/MS检测方法与流程
- 一种区分转基因玉米和非转基因玉米的检测方法与流程
- 转基因玉米事件MON87419及其使用方法与流程
- 一种快速获取转基因玉米草甘膦耐受性表型的方法与流程
- 对应于转基因事件MON89034的玉米植物和种子及其检测和使用方法与流程
- 转基因玉米品系VCO‑01981‑5检测方法和试剂与制造工艺
- 一种快速创制转基因玉米双单倍体纯合后代的方法与制造工艺