美罗培南的纯化方法与流程
本发明属于制药领域,具体涉及美罗培南的纯化方法。
背景技术:
美罗培南(meropenem),化学名(4R,5S,6S)-3-[[(3S,5S)-5-(二甲基氨甲酰基)-3-吡咯烷]硫]-6-[(1R)-1-羟乙基]-4-甲基-7-氧-1-氧双环[3,2,0]-庚-2-烯-2-羧酸三水合物,属于第二代的高效广谱的全合成碳青霉烯类抗生素。化学结构式如下:
现有技术中,美罗培南一般通过β-甲基碳青霉烯的催化氢化制备,例如使用钯碳作为催化剂,反应历程如下:
上述催化氢化的制备方法,反应得到的氢解液中,除了目的产物美罗培南,其副产物包括未完全脱除保护基的β-甲基碳青霉烯、保护基残基等各类弱极性和非极性的副产物,如果不能对这些副产物进行有效地排除,则成品药物常因杂质而存在极大的药物过敏风险。
现有技术中公开了高纯度美罗培南的制备方法(CN102372715A),但其改进点在于通过优化的催化氢化工艺条件来提高β-甲基碳青霉烯的自由基脱除率,尽管也采用了诸如萃取、结晶等常规的纯化手段,但其本质上还是通过膜分离法获得原料药级别的美罗培南,这种纯化路线成本高、产量低,在工业应用方面依然存在较大改进空间。
技术实现要素:
针对现有技术的不足,发明经过潜心研究,创造性地提出了针对美罗培南的不同极性杂质采取不同的处理方式的纯化路线,从而极大程度地简化了美罗培南的纯化工艺,在保证和提高纯度的前提下提高其产量、降低纯化成本。
具体而言,本发明所提供的美罗培南的纯化方法,包括以下步骤:
S01、准备含有美罗培南的氢解液;
S02、使用与水不溶的弱极性萃取剂对氢解液进行萃取,通过高效液相色谱法(HPLC)跟踪,以主峰面积比≧80%为萃取终点;
S03、将步骤S02的水相分离后,加入与水互溶的强极性溶剂,制备美罗培南晶体。
步骤S02中,所述萃取终点为:主峰面积比≧80%,相对保留时间(RRT)>2的总杂质峰面积比≦8%。
进一步地,所述纯化方法还包括:
S04、将步骤S03的晶体过滤收集并干燥。
步骤S01中,准备含有美罗培南的氢解液的方法没有特别限定,任何现有技术中通过催化氢化方法制备美罗培南粗品的方法,其得到的氢解液均可作为本发明的纯化方法的原料。具体操作中,可以使用β-甲基碳青霉烯为原料,以钯碳为催化剂,进行氢化催化以获得氢解液。作为优选,所述氢解液首先去除催化剂,例如可以采用过滤、离心分离等本领域常规的固液分离手段,将钯碳等催化剂去除。进一步优选在去除催化剂后进行脱色处理,例如可以向滤液中加入活性炭,震荡后过滤去除活性炭,将不含催化剂、脱色的氢解液作为步骤S02的原料。
进一步地,步骤S02中,所述与水不溶的弱极性萃取剂的介电常数≤5,具体可以选自石油醚(1.80)、正已烷(1.58)、环已烷(2.02)、氯仿(4.81)、四氯化碳(2.24)中的一种或多种组合。此萃取剂的目的在于分离去除氢解液中弱极性有机杂质,其具体的种类选择和组分配比可由本领域技术人员根据纯化所针对的氢解液的具体杂质构成来进行调节。作为优选,氢解液与水不溶的弱极性萃取剂的体积比为1:1~1.2。进一步优选萃取在10~15℃下进行,以防止温度升高对于杂质的选择性降低。萃取后,静置1~5min后分离水相和油相。
进一步地,步骤S03中,所述与水互溶的强极性溶剂的介电常数≥7,具体可以选自四氢呋喃(7.39)和/或丙酮(20.50)。次强极性溶剂一方面充当美罗培南的沉淀结晶剂,另一方面利用其强极性而将同为强极性的杂质留存于液相中,从而与晶体纯品分离。氢解液与水互溶的强极性溶剂的体积比为1:8-9,进一步优选在10~15℃下加入强极性溶剂。实际操作中,可以将与水互溶的强极性溶剂的一部分直接加入水相,再将剩余的滴加入水相,直接加入与滴加的强极性溶剂体积比优选1~2:1。
进一步地,强极性溶剂滴加完毕后,将体系温度降至5℃以促进晶体形成,养晶25-50min后,得到美罗培南晶体。
进一步地,步骤S04中,将步骤S03的含晶体混合液过滤,30~50℃下真空干燥2.5~3.5h,得到干燥的晶体。
本发明的有益效果在于:本发明创造性地一次使用弱极性溶剂和强极性溶剂,对含有美罗培南的氢解液进行纯化处理,从而得到了高纯度美罗培南原料药。该方法具有操作简单、纯化效果好、成本低廉等优点,能够应用于美罗培南的工业级纯化和生产。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面对实施例或现有技术中所需要使用的附图作简单地介绍。显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1是实施例1制备得到的美罗培南的HPLC测试结果。
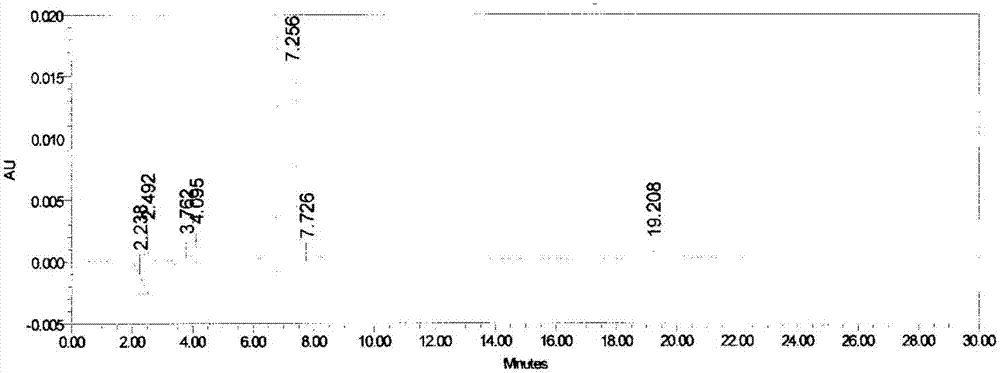
图2是实施例2制备得到的美罗培南的HPLC测试结果。
具体实施方式
下面将结合本发明的实施例,对本发明的技术方案进行清楚、完整地描述。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
在烧杯中依次加入15g保护美罗培南、400ml四氢呋喃、8g二甲基吡啶搅拌至溶清后加入到1L的氢化反应釜中,称取4g钯碳(干基)、300ml纯化水加入氢化反应釜中,密闭氢化反应釜盖。向氢化反应釜中充入氮气升压至0.3~0.35MPa,开启排空,泄压至0.10~0.15Mpa,关闭排空阀,按上述方法总共氮气置换3次,完毕后,按氮气的置换方法进行3次氢气置换。置换结束后,升氢气压力至2.0~2.2MPa、控制温度30~40℃、搅拌800r/min、反应2h。反应过程中氢气压力降低至1.5~1.6MPa时补加氢气压力至2.0~2.2MPa直至反应结束。
反应结束缓慢开启氢化釜的排空阀,排去氢气至反应釜内压力0MPa,关闭排空阀。缓慢开启氢化釜的真空阀,抽真空至-0.07MPa~-0.08MPa,真空稳定后继续抽2~3分钟。打开氮气阀,充氮气至压力0.3~0.35MPa。按以上步骤进行两次氮气置换后开釜出料,滤除钯碳。
将氢解液倒入2000ml烧杯中,控制温度10~15℃,并加入300ml四氯化碳搅拌两分钟后静置,立即分层,取样进行HPLC跟踪。HPLC跟踪采用安捷伦C18色谱柱,以0.1%三乙胺溶液:乙腈=1000:70(V/V)为流动相,在流速1.6ml/min、检测波长220nm、柱温40℃条件下进行检测。以主峰面积比≥80%,RRT>2的总杂质峰面积比≤8%为萃取终点,分出四氯化碳层回收套用,水层加入3000ml三口瓶中,控温10-15℃,一次性加入四氢呋喃1500ml,滴加四氢呋喃1000ml,水溶液浑浊有晶体析出,降温至5℃,养晶30min,过滤,-0.09kpa/40℃干燥3小时,得类白色美罗培南粗品7.7g,收率51.33%,含量99.69%。
产物的HPLC测试结果参见图1。图中,7.246min为目标产物的峰,峰面积比为99.69%。
实施例2
在烧杯中依次加入15g保护美罗培南、400ml四氢呋喃、8g二甲基吡啶搅拌至溶清后加入到1L的氢化反应釜中,称取4g钯碳(干基)、300ml纯化水加入氢化反应釜中,密闭氢化反应釜盖。向氢化反应釜中充入氮气升压至0.3~0.35MPa,开启排空,泄压至0.10~0.15Mpa,关闭排空阀,按上述方法总共氮气置换3次,完毕后,按氮气的置换方法进行3次氢气置换。置换结束后,升氢气压力至2.0~2.2MPa、控制温度30~40℃、搅拌800r/min、反应2h。反应过程中氢气压力降低至1.5~1.6MPa时补加氢气压力至2.0~2.2MPa直至反应结束。
反应结束缓慢开启氢化釜的排空阀,排去氢气至反应釜内压力0MPa,关闭排空阀。缓慢开启氢化釜的真空阀,抽真空至-0.07MPa~-0.08MPa,真空稳定后继续抽2~3分钟。打开氮气阀,充氮气至压力0.3~0.35MPa。按以上步骤进行两次氮气置换后开釜出料,滤除钯碳。
将氢解液倒入2000ml烧饼中,控制温度10~15℃,并加入300ml石油醚搅拌两分钟后静置分层,取样进行HPLC跟踪。HPLC跟踪采用安捷伦C18色谱柱,以0.1%三乙胺溶液:乙腈=1000:70(V/V)为流动相,在流速1.6ml/min、检测波长220nm、柱温40℃条件下进行检测。以主峰面积比≧80%,RRT>2的总杂质峰面积比≦8%为萃取终点,分出石油醚层回收套用,水层加入3000ml三口瓶中,控温10-15℃,一次性加入丙酮1500ml,滴加丙酮1000ml,水溶液浑浊有晶体析出,降温至5℃,养晶30min,过滤,-0.09kpa/40℃干燥3小时,得类白色美罗培南粗品7.72g,收率51.47%,含量99.64%。
产物的HPLC测试结果参见图2。图中,7.256min为目标产物的峰,含量为99.64%。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!