一种聚合物基纳米凝胶及制备方法与应用与流程
本发明涉及重金属检测材料领域,尤其涉及一种聚合物基纳米凝胶及制备方法与应用。
背景技术:
重金属污染能通过食物链传递,对人体产生有害甚至是致命的影响。其中,以汞离子对人体的损害尤为突出,汞离子进入人体后所形成的甲基汞具有神经毒性,会导致脑损伤、各种认知和运动障碍。开发快速、便捷的重金属离子检测和清除的方法对于维护公共安全具有重要意义。
罗丹明b是一种以氧杂蒽大刚性共轭平面为发色团的碱性荧光染料,荧光量子产率高、光稳定性好、波长范围宽,常用于各种离子、多肽、酶、蛋白质和遗传物质的分析检测。基于罗丹明b改性而来的探针检测原理一般是建立在衍生物中螺内酰胺的闭环-开环转变过程上:在中性或碱性条件下,罗丹明b衍生物呈现出闭环结构,无可见光吸收、无荧光;但当检测体系呈酸性或出现目标离子或小分子时,罗丹明b衍生物转变成开环结构,在可见光区域出现很强的吸收,探针溶液转变为玫红色,并产生很强的红色荧光。迄今为止,已有多例罗丹明b衍生物被成功应用于汞离子检测,但是绝大多数探针水溶性差,不能在100%纯水环境中实现检测,必须要在有机-水混合溶剂中才能进行(如甲醇水溶液,乙腈水溶液等)。
因此,现有技术还有待于改进和发展。
技术实现要素:
鉴于上述现有技术的不足,本发明的目的在于提供一种聚合物基纳米凝胶及制备方法与应用,旨在解决现有的hg2+检测、去除材料无法应用于纯水环境中的问题。
本发明的技术方案如下:
一种聚合物基纳米凝胶的制备方法,包括如下:
步骤a、将nipam、oegma、表面活性剂和交联剂在保护气氛中溶于去离子水,然后加热至60-80℃,并加入引发剂,混合均匀;
步骤b、将含rhbha和nba的有机溶剂溶液,加入到所述步骤a中的混合溶液中,反应7-10h,然后提纯处理,得到聚合物基纳米凝胶p(nipam-co-oegma-co-nba-co-rhbha)。
所述的聚合物基纳米凝胶的制备方法,其中,相对于所述聚合物基纳米凝胶的体积,所述表面活性剂的用量为1~5mmol/l。
所述的聚合物基纳米凝胶的制备方法,其中,相对于所述聚合物基纳米凝胶的体积,所述交联剂的用量为1~5mmol/l。
所述的聚合物基纳米凝胶的制备方法,其中,相对于所述聚合物基纳米凝胶的体积,所述引发剂的用量为2~4mmol/l。
所述的聚合物基纳米凝胶的制备方法,其中,所述步骤b中,提纯处理包括步骤:先抽滤除去杂质,然后采用截留分子量为1000的透析袋透析。
一种聚合物基纳米凝胶,采用如上所述的制备方法制备而成。
所述的聚合物基纳米凝胶,包括如下重量百分比的组分:
nipam70~89%,
oegma5~15%,
nba5~15%,
rhbha0.5%-5%。
一种如上所述的聚合物基纳米凝胶的应用,将所述聚合物基纳米凝胶用于检测hg2+。
一种如上所述的聚合物基纳米凝胶的应用,将所述聚合物基纳米凝胶用于去除hg2+。
所述的聚合物基纳米凝胶的应用,其中,将所述聚合物基纳米凝胶用于去除hg2+的方法中,包括:采用uv光照射。
有益效果:本发明提供了一种如上所述的聚合物基纳米凝胶的制备方法,将hg2+响应性荧光探针单体丙烯酸硫脲酯(rhbha)和光响应性单体邻硝基苄基丙烯酸酯(nba),通过自由基乳液聚合方法共价修饰到亲水基体聚n-异丙基丙烯酰胺(pnipam)和寡聚乙二醇甲醚甲基丙烯酸酯(oegma)构成的聚合物纳米凝胶基体p(nipam-co-oegma)中,利用p(nipam-co-oegma)以及rhbha所带的硫脲基团的亲水性,本发明的聚合物基纳米凝胶能在纯水中选择性地识别hg2+,所得的纳米凝胶中的rhbha可以与hg2+相互作用,并且抗干扰性强,检测灵敏度高,室温下的检测限为2.37×10-8mol/l;进一步利用nba的光响应性,纳米凝胶在紫外(例如365nm)光照射下,nba的邻硝基苄基断裂,从而释放出聚丙烯酸(paa),螯合水溶液中的hg2+,随着光照时间的不同,nba的降解程度也不同,可通过光照时间调节体系中paa的含量,实现hg2+的按需清除。
附图说明
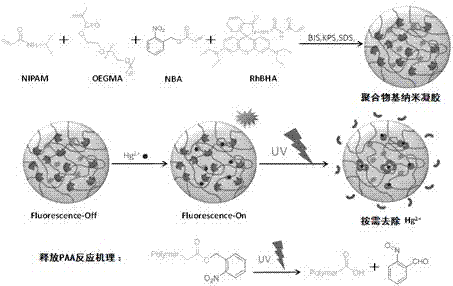
图1为p(nipam-co-oegma-co-nba-co-rhbha)的反应原理及检测、去除hg2+的原理图。
图2a为清除剂邻硝基苄基丙烯酸酯单体(nba)的1h-nmr。
图2b为清除剂邻硝基苄基丙烯酸酯单体(nba)的13c-nmr。
图3a为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的sem图(低倍数)。
图3b为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的sem图(高倍数)。
图4为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的eds全谱分析图。
图5a为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的eds能谱图-分层图像。
图5b为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的eds能谱图-c图像。
图5c为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的eds能谱图-o图像。
图5d为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的eds能谱图-s图像。
图6为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶加入hg2+后可见吸收和荧光发射谱的变化。
图7为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶加入不同金属阳离子的荧光谱图。
图8a为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶(0.05g/l)随hg2+浓度变化(0-10μm)的荧光谱图。
图8b为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶(0.05g/l)随hg2+浓度变化(0-10μm)的在λem=590nm荧光强度变化。
图9a为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶(0.05g/l)随hg2+浓度变化(0-100μm)的可见吸收光谱。
图9b为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶(0.05g/l)随hg2+浓度变化的在λ=568nm的吸光度变化。
图10为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的抗干扰性试验结果图。
图11a为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶(0.05g/l)在不同ph条件下的荧光发射光谱。
图11b为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶(0.05g/l)在不同ph条件下,在λem=587nm荧光变化曲线。
图12为nba单体光响应的吸收光谱。
图13a为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶随着紫外照射时间的光响应性谱图。
图13b为p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶随着紫外照射时间的不同在309nm处的吸收变化。
图14a为p(nipam-co-oegma-co-nba-co-rhbha)在4μm[hg2+]下纳米凝胶随着紫外照射时间的光响应性谱图。
图14b为p(nipam-co-oegma-co-nba-co-rhbha)在4μm[hg2+]下纳米凝胶随着紫外照射时间的不同在309nm处的吸收变化。
具体实施方式
本发明提供了一种聚合物基纳米凝胶及制备方法与应用,为使本发明的目的、技术方案及效果更加清楚、明确,以下对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
本发明提供的一种聚合物基纳米凝胶的制备方法包括如下:
步骤a、将nipam、oegma、表面活性剂和交联剂在保护气氛中溶于去离子水,然后加热至60-80℃,并加入引发剂,混合均匀;
步骤b、将含rhbha和nba的有机溶剂溶液,加入到所述步骤a中的混合溶液中,反应7-10h,然后提纯处理,得到聚合物基纳米凝胶p(nipam-co-oegma-co-nba-co-rhbha)。
作为上述方法的一种较为优选的实施方式,具体的,将nipam、oegma,表面活性剂乳化剂十二烷基硫酸钠(sds),交联剂n,n-亚甲基双丙烯酰胺(bis)溶于去离子水,通氮气持续搅拌均匀;逐渐加热升温至60-80℃,将引发剂过硫酸钾(kps)溶在去离子水中,然后将其加入反应体系中,混合均匀;再依次将汞离子荧光探针rhbha和邻硝基苄基丙烯酸酯nba分别溶在四氢呋喃中,并缓慢(例如滴加)加入到反应体系中,共聚反应7-10h,最后提纯处理,得到聚合物基纳米凝胶p(nipam-co-oegma-co-nba-co-rhbha),制备原理参照图1。
本发明将hg2+响应性荧光探针单体丙烯酸硫脲酯(rhbha)和光响应性单体邻硝基苄基丙烯酸酯(nba),通过自由基乳液聚合方法共价修饰到亲水基体聚n-异丙基丙烯酰胺(pnipam)和寡聚乙二醇甲醚甲基丙烯酸酯(oegma)构成的聚合物纳米凝胶基体中,所得的纳米凝胶中的rhbha可以与hg2+相互作用,并且纳米凝胶能够在纯水中选择性地识别hg2+,抗干扰性强,检测灵敏度高,室温下的检测限为2.37×10-8mol/l;进一步利用nba的光响应性,纳米凝胶在紫外(例如365nm)光照射下,nba的邻硝基苄基断裂,从而释放出聚丙烯酸(paa),螯合水溶液中的hg2+,随着光照时间的不同,nba的降解程度也不同,可通过光照时间调节体系中paa的含量,实现hg2+的按需清除。
优选的,相对于所述聚合物基纳米凝胶的体积,所述表面活性剂的用量为1~5mmol/l,超出范围使得生成的纳米凝胶中的球状颗粒太小,性能降低。
优选的,相对于所述聚合物基纳米凝胶的体积,所述交联剂的用量为1~5mmol/l,超出范围会使得纳米凝胶中的交联点过多,链段活动力变差。
优选的,相对于所述聚合物基纳米凝胶的体积,所述引发剂的用量为2~4mmol/l,太少会极大降低反应效率,太多则会形成小分子量的纳米凝胶,不能起到有效的作用。
优选的,提纯处理包括步骤:先抽滤除去杂质,然后采用截留分子量为1000的透析袋透析,去除表面活性剂、引发剂等小分子,纯化纳米凝胶。具体可采用去离子水透析4天,水每天更换4-6次。
本发明还提供了一种聚合物基纳米凝胶,采用上述制备方法制备而成。本发明的聚合物基纳米凝胶能选择性地识别hg2+,抗干扰性强,检测灵敏度高,并且可以按需去除hg2+,兼具检测和去除hg2+功能,可应用于溶剂为纯水的环境中。
优选的,所述的聚合物基纳米凝胶,包括如下重量百分比的组分:
nipam70~89%,
oegma5~15%,
nba5~15%,
rhbha0.5%-5%。
本发明还提供了一种如上所述的聚合物基纳米凝胶的应用,将所述聚合物基纳米凝胶用于检测hg2+和/或去除hg2+,具体原理可参照图1。
优选的,将所述聚合物基纳米凝胶用于去除hg2+时:采用uv光(例如波长为365nm)照射,通过控制光照时间长度,来调节nba中释放出的paa数量,达到按需去除hg2+。
下面通过实施例对本发明进行详细说明。
实施例1
(1)邻硝基苄基丙烯酸酯nba的合成
称取邻硝基苄醇(10g,65.35mmol)和三乙胺et3n(9.3ml,65.43mmol)装入干燥的两口瓶中,再加入60ml的无水ch2cl2,在磁力搅拌下,冰水浴30min,再用适量的无水ch2cl2稀释丙烯酰氯,通过滴液漏斗缓慢滴加入两口瓶中。
滴加完毕后,撤掉冰水浴,让混合物恢复至室温,继续搅拌24h,关掉反应,用去离子水进行分液洗涤,除去杂质。然后用无水mgso4进行干燥,抽滤,用旋转蒸发仪除去所有溶剂,得一黄棕色粘性液体。
最后,将黄棕色粗成品在硅胶色谱柱上提纯,洗脱剂石油醚和乙酸乙酯的比例为3:1,所收集成分通过旋转蒸发仪浓缩,得到粘性无色液体,得到核磁谱图如图2a和图2b。
(2)p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的制备
将n-异丙基丙烯酰胺nipam(0.4526g,4mmol),寡聚乙二醇甲醚甲基丙烯酸酯oegma(0.25g,0.5mmol),表面活性剂乳化剂十二烷基硫酸钠sds(0.0430g,0.15mmol),交联剂n,n-亚甲基双丙烯酰胺bis(0.0153g,0.1mmol)溶于去离子水(50ml),通氮气持续搅拌30min。
逐渐加热升温至70℃,将引发剂过硫酸钾kps(0.0405g,0.15mmol)溶在4.0ml的去离子水中,用试管将其加入反应体系中。分别称取汞离子荧光探针rhbha(0.0285g,0.05mmol)溶在8ml的四氢呋喃中,邻硝基苄基丙烯酸酯nba(0.1035g,0.5mmol)溶在4ml四氢呋喃中,并一滴一滴缓慢加入到反应体系中。
维持反应温度继续共聚8h,冷却至室温后,用布氏漏斗抽滤除去杂质,最后再将所得粗产物注入截留分子量为1000的透析袋,再用去离子水透析4天(水要每天更换4-6次),从而获得纳米凝胶产物。
下面结合附图对该检测材料和检测结果做进一步说明。
(1)p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的形貌
合成的p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的形貌如图3a和图3b所示,可以看出干态纳米凝胶主要呈完整球状结构,均匀分散,粒径大小约为100-200nm,在放大倍数的图中也可看到大小较为均匀。
(2)p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的eds元素分析
如面总谱图4所示,由于是将纳米凝胶滴在硅片上做的扫描测试,因此si元素的的含量最高。另外,不难发现,eds的谱图中依次出现了c、o、s这三种元素的谱峰,主要来源于聚合物基体和rhbha单体。由面分布能谱图5a、图5b、图5c和图5d可知,存在的c、o、s三种元素主要集中分布在纳米凝胶区域,其中以作为聚合物骨架的c元素最多,o、s元素分别次之,这均证明了rhbha被成功引入聚合物基纳米凝胶中。
(3)p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的hg2+检测选择性
由图6可看出,未加入汞离子前,在400-750nm范围内,纳米凝胶体系会显示较弱的荧光强度和吸光度,加入汞离子后,在568nm和590nm处吸光度和荧光发射强度有较大增强。
在试管中分别加入20μl合成的纳米凝胶溶液,16μl10-3m的各种金属离子溶液,用去离子水定容至4000μl,振荡均匀后,测得荧光光谱如图7所示。相同的测试条件下(激发波长λex=500nm,狭缝宽度:ex.5nm,em.5nm),在纯纳米凝胶体系(0.05g/l)分别加入na+、k+、ca2+、co2+、zn2+、ni2+、cr3+、cu2+、cd2+、al3+、fe2+、fe3+、pb2+、hg2+、mg2+共15种常见的金属阳离子(4μm)时,只有hg2+可以使得纳米凝胶荧光强度显著增强,而铜离子则引起体系荧光猝灭,剩余的其他金属离子检测体系的荧光强度均无发生明显变化,这充分证明了纳米凝胶对hg2+的检测具有单一选择性。
(4)p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的hg2+荧光检测灵敏度
在试管中分别加入20μl合成的纳米凝胶溶液,再加入不同浓度的汞离子溶液,用去离子水定容至4000μl,混合均匀后,用来待测荧光和吸收光谱。
如图8a,25℃纯水下,随着hg2+浓度的增加,纳米凝胶(0.05g/l)的荧光强度逐渐增强,并伴随着微弱的红移。当hg2+浓度达到4μm以后,荧光强度趋于饱和。图8b显示,当汞离子浓度为0-3μm,纳米凝胶对hg2+的荧光响应强度呈线性快速增长趋势。利用三倍噪音法,测出纳米凝胶的线性段增长斜率为74.749,空白样的标准方差为0.590,计算测得该纳米凝胶在25℃下的检出限为2.37×10-8m。
纳米凝胶(0.05g/l)检测汞离子的可见吸收光谱如图9a,无hg2+存在的空白体系,纳米凝胶在568nm处的吸光度为0.7,而随着hg2+浓度逐渐增大(0-100μm),纳米凝胶的吸光度不断增强,当汞离子浓度达到40μm以后,吸光度达到最大值2.1。如图9b,在汞离子浓度为0-40μm,纳米凝胶的吸光度呈类线性式的增长,直到40μm以后,吸光度达到饱和,形成稳定平台。
(5)p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的抗干扰性
在试管中分别加入20μl合成的纳米凝胶溶液,16μl10-3m的汞离子溶液,16μl10-3m的其他金属离子溶液,用去离子水定容至4000μl,振荡均匀后,测得荧光光谱。设置仪器参数:激发波长λex=500nm,狭缝宽度:ex.5nm,em.5nm。
研究共存离子对p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶检测hg2+的干扰如图10所示,当hg2+(4μm)分别与na+、k+、ca2+、co2+、zn2+、ni2+、cr3+、cu2+、cd2+、al3+、fe2+、fe3+、pb2+、mg2+共14种离子(4μm)共存时,除了铜离子由于自身d9结构的顺磁性会使纳米凝胶溶液(0.05g/l)荧光强度趋于猝灭,其余的金属阳离子共混体系的荧光强度均显著增强,且与单独检测hg2+的体系强度大小差异甚微,由此可知干扰离子的加入没有影响到该纳米凝胶对汞离子的检测性能。另外,当用纳米凝胶分别检测15种金属离子时,hg2+检测体系引起的荧光强度明显高于其余14种金属离子体系的强度,直观证明了该纳米凝胶对hg2+的选择性。
(6)p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的ph响应性
在试管中加入20μl合成的纳米凝胶溶液,再用盐酸和氢氧化钠配制不同ph值(ph1-14)的水溶液,并取3980μl所配的不同ph溶液定容至4000μl,振荡均匀后,用ph计测量最终ph值,用来待测荧光光谱。设置仪器参数:激发波长λex=500nm,狭缝宽度:ex.5nm,em.5nm。
纳米凝胶ph响应行为如图11a所示,当体系中ph值(0.61-13.77)变化时,纳米凝胶在587nm处的荧光强度逐渐减弱,并伴随着微弱的蓝移。这主要是由于酸性条件下,质子化作用诱导了螺内酰胺闭环转变为开环状态,从而使荧光强度相较于中性或碱性体系有所增强。图11b,在ph值为0-6范围,该纳米凝胶的荧光强度由300降至200,下降了约33%左右;当ph值由6-14变化时,荧光强度持续下降,可能是氢氧化钠调节ph所引起的离子浓度增大所致。
(7)nba单体的光响应性
nba单体的光诱导降解行为如图12所示,无365nm紫外光照时,nba单体在265nm处出现特征吸收峰,归属于邻硝基苄酯基;而在uv光照后,265nm处的吸收峰消失,而在309nm处出现吸收峰,主要是因为nba的邻硝基苄基出现化学键断裂,导致亚硝基苯甲醛中醛基的生成。
(8)p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶的光响应性
纳米凝胶p(nipam-co-oegma-co-nba-co-rhbha)的光响应行为如图13a所示,无紫外光照前,p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶溶液在263nm处有一特征吸收峰,归属于纳米凝胶中nba邻硝基苄酯的酯基。随着光照时间的延长,263nm处的吸光度逐渐降低,伴随着309nm处吸收峰的出现和逐渐升高,主要因为nba分子的断裂,导致酯基逐渐消失,醛基逐渐生成,直到最后断裂完全,吸光度不再变化。且图13b显示,随着光照时间的增加,纳米凝胶在309nm处的吸光度几乎呈类线性增强,持续160min后,吸光度达到稳定,不再变化。这也侧面证明了纳米凝胶中nba的光裂解速率几乎维持为一恒定值,直到最后所有nba完全裂解为止。
而在hg2+存在时,p(nipam-co-oegma-co-nba-co-rhbha)纳米凝胶在汞离子存在的体系,如图14a所示,光照前,纳米凝胶在265nm处仍有吸收峰,而随着光照时间的延长,265nm处的吸收峰缓慢降低,而在309nm处出现吸收峰,并缓慢升高,这与无汞离子存在的纳米凝胶的光响应行为是一致的。其断裂的速率如图14b所示,在hg2+存在时,纳米凝胶在309nm处的吸光度增长几乎呈匀速变化,当光照时间达210min后,吸光度达到饱和。相较于无hg2+的体系,纳米凝胶中nba完全断裂所需要的光照时间要长些,可能是因为汞离子的离子浓度对光作用的干扰所致。
(9)纳米凝胶对hg2+的清除效果
在试管中分别加入1600μl纳米凝胶溶液,160010-4m的汞离子溶液,再用去离子水定容至8000μl,从而得到测试样品。配制两个相同的样品,分别用uv光照150min和250min,用摇床振荡30min,再注入截留分子量为1000的透析袋,用200ml去离子水透析18h,待吸附平衡后,取透析清液进行icp-aes的汞离子浓度测试,从而求得透析袋对汞的截留量。清除率则通过平衡后透析袋对汞的截留量除以汞的总投入量而得。
纳米凝胶(2.04g/l)在hg2+溶液(4mg/l)中对hg2+的清除效果如表1,用365nm紫外光持续照射体系150min后,icp-aes测得透析清液hg2+浓度为28.6mg/ml,计算得纳米凝胶对汞离子的清除率为82.13%;随着光照时间的延长至250min,测得透析清液hg2+浓度为4.9mg/ml,清除率达到96.89%。可看出,在清除剂没完全断裂之前,相同测试条件下,延长光照时间,可以提高体系中对hg2+的清除率。纳米凝胶对hg2+的按需清除机理:uv辐射导致nba邻硝基苄酯基断裂,生成聚丙烯酸paa,以paa作为重金属离子的螯合剂,清除hg2+。固定光强,延长uv辐射时间,提高nba降解程度,从而增加清除体系中paa的含量,以络合更多的hg2+,提高汞离子的清除率。同时,由于paa所带有的羧基具有亲水性,也能提高paa与水溶液中hg2+的碰撞效率,增强清除效果。因此,根据环境体系hg2+含量,通过改变紫外光照射纳米凝胶的时间,以控制paa的生成量,可以实现hg2+可控按需清除的功能。
综上所述,本发明提供了一种聚合物基纳米凝胶及制备方法与应用,本发明将hg2+响应性荧光探针单体丙烯酸硫脲酯(rhbha)和光响应性单体邻硝基苄基丙烯酸酯(nba),通过自由基乳液聚合方法共价修饰到亲水基体聚n-异丙基丙烯酰胺(pnipam)和寡聚乙二醇甲醚甲基丙烯酸酯(oegma)构成的聚合物纳米凝胶基体中,所得的纳米凝胶能够在纯水中选择性地识别hg2+,并实现对hg2+的按需清除。
应当理解的是,本发明的应用不限于上述的举例,对本领域普通技术人员来说,可以根据上述说明加以改进或变换,所有这些改进和变换都应属于本发明所附权利要求的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!