一种一锅法制备1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮的方法与流程
本发明属于有机合成技术领域,具体涉及一种一锅法制备1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮的方法。
背景技术:
1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮的分子结构式:
它是一种β-二酮类化合物,这是一类含有1,3-二羰基结构的化合物,分子内存在酮式-烯醇式之间的异构化转变,其具有高的吸收系数,对稀土离子有很强的配位能力,与稀土离子配位形成稳定的鳌合六元环,敏化稀土离子发光。由于其高发光效率及稳定的物理、化学性质,使得其在分析化学、稀土转光薄膜、太阳能电池光转换胶膜、防伪、光致发光和电致发光等方面有着宽广的应用前景。1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮还是合成3-(4-苯甲酰氨基-苯基)-5-苯基吡唑(cas:112248-32-5)等杂环化合物的重要中间体。
1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮是一种含有苯甲酰氨基的β-二酮,程果等人的文献:新的稀土eu,tb(iii)β-二酮三元配合物的合成与光谱性质中,4-氨基苯乙酮和苯甲酸酯在碱性条件下经过克莱森缩合反应,以56.48%的收率得到1-(4-氨基苯基)-3-苯基丙烷-1,3-二酮;任晓明的博士学位论文:稀土铕、铽有机配合物的制备与性能研究中,4-氨基苯乙酮和苯甲酸酯在碱性条件下经过克莱森缩合反应,以53.56%的收率得到1-(4-氨基苯基)-3-苯基丙烷-1,3-二酮;两篇文献均没有制得1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮。hauser等人通过两步法制备了1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮(thejournaloforganicchemistry,1957,22(8):909-912.),但是后处理需要加入铜离子螯合,螯合物再经浓硫酸酸化、有机溶剂萃取,才制得1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮,收率仅为32%~56%,后处理繁琐,收率低。
技术实现要素:
为解决现有技术中制备方法及后处理的繁琐以及产物收率低的问题,本发明提供一种一锅法制备1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮的方法。
4-氨基苯乙酮中的对位氨基与苯甲酸酯在碱性条件下酰胺化形成中间体n-(4-乙酰基苯基)苯甲酰胺,该中间体继续与苯甲酸酯发生克莱森缩合反应,制得1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮。具体通过以下技术方案一锅法实现:
惰性气体保护下,将4-氨基苯乙酮于冰水浴5℃以下,加入催化剂,搅拌下滴加苯甲酸酯的四氢呋喃溶液,滴加时间为1~2小时,滴加完毕撤去冰水浴,升至室温,加热并保温反应5~10小时,待原料4-氨基苯乙酮以及中间体n-(4-乙酰基苯基)苯甲酰胺反应完全后,调节反应液ph值为6.5~7.5,分液、盐洗有机层、旋蒸;有机溶剂重结晶、过滤干燥后制得淡黄色固体1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮。
为进一步描述制备1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮的过程,合成反应路线如下:
进一步地,所述4-氨基苯乙酮、催化剂、苯甲酸酯按物质的量之比为1:(2.1~2.6):(2.1~3.0)。
优选地,所述4-氨基苯乙酮、催化剂、苯甲酸酯按物质的量之比为1:2.2:2.5。
进一步地,所述保温反应是在50~60℃下进行的;温度若低于50℃,反应时间较长,温度若高于60℃,反应液颜色加深,不利于后处理。
进一步地,所述调节反应液ph值是通过质量分数为12%~20%的盐酸来调节。
进一步地,所述苯甲酸酯为苯甲酸甲酯或苯甲酸乙酯。
优选地,所述苯甲酸酯为苯甲酸甲酯。
进一步地,所述催化剂为氢化钠、氢化钾、乙醇钠、叔丁醇钠、乙醇钾、叔丁醇钾中的一种。
进一步地,所述中间体n-(4-乙酰基苯基)苯甲酰胺无需进行分离,一步合成1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮。
本发明的有益效果是:本发明方法一锅实现制备1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮。4-氨基苯乙酮中的对位氨基与苯甲酸酯在碱性条件下优先发生酰胺化反应形成中间体n-(4-乙酰基苯基)苯甲酰胺,中间体无需分离,直接继续与苯甲酸酯发生克莱森缩合反应,最终制得1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮。本发明方法一锅合成产物,无需分离中间体就能得到高收率产物,简化操作的同时减少废水废渣的产生,大大降低成本,本发明方法制得的产物纯度高、收率高,产物收率稳定在85%以上。
附图说明
图1是中间体n-(4-乙酰基苯基)苯甲酰胺的核磁共振氢谱图。
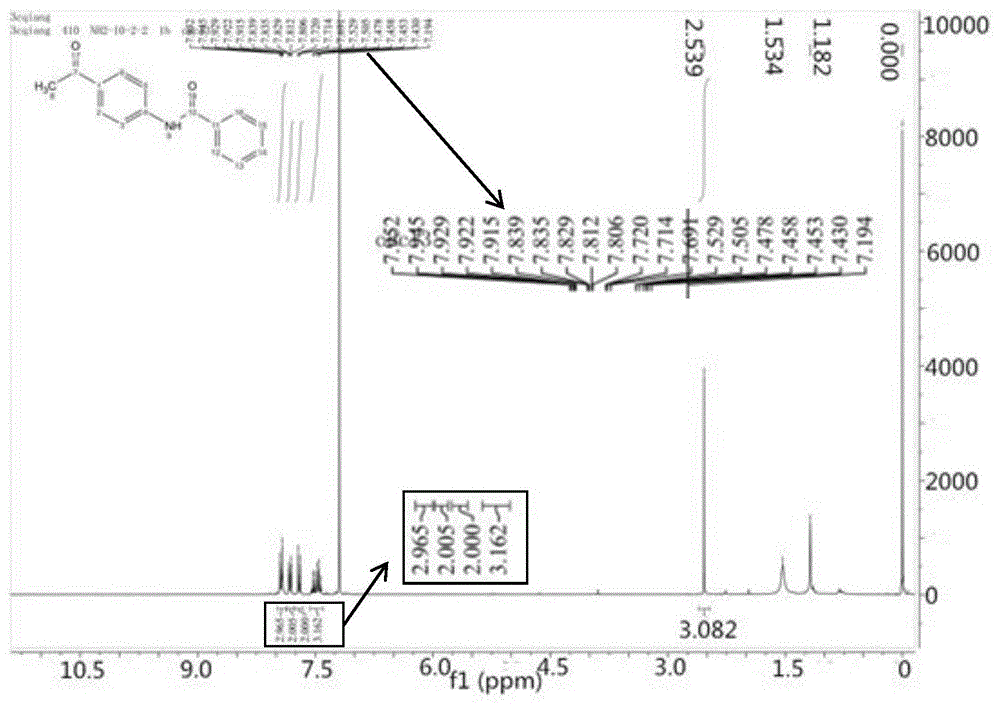
图2是实施例1产物1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮的核磁共振氢谱图。
具体实施方式
以下结合具体实施例进一步描述本发明,但不限制本发明范围。
实施例1
预先将所有仪器放烘箱内83℃干燥2h,氮气保护下,将33.79g(0.25mol)的4-氨基苯乙酮溶于盛有800ml干燥四氢呋喃的三口烧瓶中,冰水浴降温至5℃以下,搅拌下分批将22.0g(0.55mol)nah(60%dispersioninmineraloil)加入到反应瓶中,搅拌30min,溶液呈灰色,出现少量气泡,无明显升温;将85.77g(0.63mol)苯甲酸甲酯溶于250ml干燥的四氢呋喃中,转移至恒压滴液漏斗并向三口烧瓶中缓慢滴加,1.5h滴加完毕,撤去冰水浴,升温至55℃进行保温反应,随着反应进行,反应液逐渐变成土黄色,薄层层析(tlc)跟踪反应情况,2.5h后4-氨基苯乙酮反应完全,继续tlc跟踪反应情况,5.5h后中间体n-(4-乙酰基苯基)苯甲酰胺反应完全,停止反应;用15%稀盐酸调节溶液ph值为7.0,溶液分层,100ml四氢呋喃萃取下层水相,合并有机层,饱和食盐水洗50ml×2、无水硫酸钠干燥有机相2h,抽滤旋蒸棕黄色固体,石油醚清洗除去矿物油,所得固体用乙醇重结晶得淡黄色固体产物1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮75.8g,产率为88.3%。
将中间体及产物收集用于核磁共振的表征,中间体的核磁共振谱图如图1所示,由图1可知,生成的中间体确是n-(4-乙酰基苯基)苯甲酰胺;产物的核磁共振谱图如图2所示,由图2可知,制得的产物确是1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮。测得产物熔点为183.1~183.7℃。
实施例2
预先将所有仪器放烘箱内83℃干燥2h,氮气保护下,将33.79g(0.25mol)的4-氨基苯乙酮溶于盛有1000ml干燥四氢呋喃的三口烧瓶中,冰水浴降温至5℃以下,搅拌下分批将21.25g(0.53mol)kh(30%dispersioninmineraloil)加入到反应瓶中,搅拌30min,溶液呈灰色,出现少量气泡,无明显升温;将72.16g(0.53mol)苯甲酸甲酯溶于250ml干燥的四氢呋喃中,转移至恒压滴液漏斗并向三口烧瓶中缓慢滴加,1.5h滴加完毕,撤去冰水浴,升温至50℃进行保温反应,随着反应进行,反应液逐渐变成土黄色,薄层层析(tlc)跟踪反应情况,2.0h后4-氨基苯乙酮反应完全,继续tlc跟踪反应情况,6.0h后中间体n-(4-乙酰基苯基)苯甲酰胺反应完全,停止反应;用12%稀盐酸调节溶液ph值为6.5,溶液分层,100ml四氢呋喃萃取下层水相,合并有机层,饱和食盐水洗50ml×2、无水硫酸钠干燥有机相2h,抽滤旋蒸的棕黄色固体,石油醚清洗除去矿物油,所得固体用乙醇重结晶得淡黄色固体1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮71.0g,产率为85.1%。测得产物熔点为182.8~183.6℃。
实施例3
预先将所有仪器放烘箱内83℃干燥2h,氮气保护下,将33.79g(0.25mol)的4-氨基苯乙酮溶于盛有800ml干燥四氢呋喃的三口烧瓶中,冰水浴降温至5℃以下,搅拌下分批将40.83g(0.6mol)乙醇钠加入到反应瓶中,搅拌30min,溶液呈淡黄色,出现少量气泡,无明显升温;将78.97g(0.58mol)苯甲酸甲酯溶于250ml干燥的四氢呋喃中,转移至恒压滴液漏斗并向三口烧瓶中缓慢滴加,1.5h滴加完毕,撤冰水浴,升温至60℃进行保温反应,随着反应进行,反应液逐渐变成土黄色,薄层层析(tlc)跟踪反应情况,2.5h后4-氨基苯乙酮反应完全,继续tlc跟踪反应情况,6.5h后中间体n-(4-乙酰基苯基)苯甲酰胺反应完全,停止反应;用17%稀盐酸调节溶液ph值为7.0,溶液分层,150ml四氢呋喃萃取下层水相,合并有机层,饱和食盐水洗50ml×2、无水硫酸钠干燥有机相2h,抽滤旋蒸的土黄色固体,用乙醇重结晶得淡黄色1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮固体75.2g,产率为87.6%。测得产物熔点为183.0~183.6℃。
实施例4
预先将所有仪器放烘箱内83℃干燥2h,氮气保护下,将33.79g(0.25mol)的4-氨基苯乙酮溶于盛有1000ml干燥四氢呋喃的三口烧瓶中,冰水浴降温至5℃以下,搅拌下分批将54.7g(0.65mol)乙醇钾加入到反应瓶中,搅拌30min,溶液呈淡黄色,出现少量气泡,无明显升温;将94.61g(0.63mol)苯甲酸乙酯溶于250ml干燥的四氢呋喃中,转移至恒压滴液漏斗并向三口烧瓶中缓慢滴加,1.5h滴加完毕,撤冰水浴,升温至52℃进行保温反应,随着反应进行,反应液逐渐变成土黄色,薄层层析(tlc)跟踪反应情况,3.0h后4-氨基苯乙酮反应完全,继续tlc跟踪反应情况,6.5h后中间体n-(4-乙酰基苯基)苯甲酰胺反应完全,停止反应;用20%稀盐酸调节溶液ph值为6.5,溶液分层,150ml四氢呋喃萃取下层水相,合并有机层,饱和食盐水洗50ml×2、无水硫酸钠干燥有机相2h,抽滤旋蒸的土黄色固体,用乙醇重结晶得淡黄色1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮固体74.8g,产率为87.1%。测得产物熔点为182.9~183.5℃。
实施例5
预先将所有仪器放烘箱内83℃干燥2h,氮气保护下,将33.79g(0.25mol)的4-氨基苯乙酮溶于盛有1000ml干燥四氢呋喃的三口烧瓶中,冰水浴降温至5℃以下,搅拌下分批将52.85g(0.55mol)叔丁醇钠加入到反应瓶中,搅拌30min,溶液呈淡黄色,出现少量气泡,无明显升温;将95.31g(0.7mol)苯甲酸甲酯溶于250ml干燥的四氢呋喃中,转移至恒压滴液漏斗并向三口烧瓶中缓慢滴加,1.5h滴加完毕,撤冰水浴,升温至55℃进行保温反应,随着反应进行,反应液逐渐变成土黄色,薄层层析(tlc)跟踪反应情况,2.5h后4-氨基苯乙酮反应完全,继续tlc跟踪反应情况,7.0h后中间体n-(4-乙酰基苯基)苯甲酰胺反应完全,停止反应;用15%稀盐酸调节溶液ph值为7.5,溶液分层,150ml四氢呋喃萃取下层水相,合并有机层,饱和食盐水洗50ml×2、无水硫酸钠干燥有机相2h,抽滤旋蒸的土黄色固体,用乙醇重结晶得淡黄色1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮固体74.6g,产率为86.9%。测得产物熔点为183.0~183.8℃。
实施例6
预先将所有仪器放烘箱内83℃干燥2h,氮气保护下,将33.79g(0.25mol)的4-氨基苯乙酮溶于盛有1000ml干燥四氢呋喃的三口烧瓶中,冰水浴降温至5℃以下,搅拌下分批将61.71g(0.55mol)叔丁醇钾加入到反应瓶中,搅拌30min,溶液呈淡黄色,出现少量气泡,无明显升温;将112.63g(0.75mol)苯甲酸乙酯溶于250ml干燥的四氢呋喃中,转移至恒压滴液漏斗并向三口烧瓶中缓慢滴加,1.5h滴加完毕,撤冰水浴,升温至58℃进行保温反应,随着反应进行,反应液逐渐变成土黄色,薄层层析(tlc)跟踪反应,2.5h后4-氨基苯乙酮反应完全,继续tlc跟踪反应,6.5h后中间体n-(4-乙酰基苯基)苯甲酰胺反应完全,停止反应;用15%稀盐酸调节溶液ph值为7.0,溶液分层,150ml四氢呋喃萃取下层水相,合并有机层,饱和食盐水洗50ml×2、无水硫酸钠干燥有机相2h,抽滤旋蒸的土黄色固体,用乙醇重结晶得淡黄色1-(4-苯甲酰氨基-苯基)-3-苯基丙烷-1,3-二酮固体74.7g,产率为87.01%。测得产物熔点为182.8~183.5℃。
- 还没有人留言评论。精彩留言会获得点赞!