一种3-环丙基环丁酮的制备方法与流程
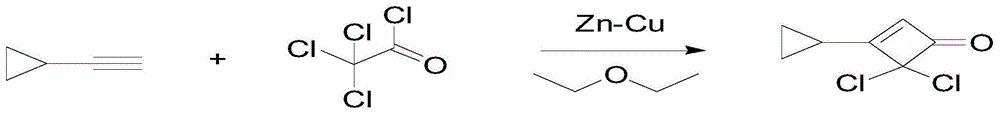
本发明属于有机合成技术领域,更具体地说,它涉及一种3-环丙基环丁酮的制备方法。
背景技术:
3-环丙基环丁酮是一种重要的化工中间体,其分子式为c7h10o,其摩尔质量为110g/mol,结构式为
现有文献报道的合成3-环丙基环丁酮如下:
此方法所用的起始原料乙烯基环丙烷市场上没有销售,所以此方法主要问题是原料获到困难,而我们的合成路线中起始原料为环丙基乙炔为工业化产品,市场易于购买,价格低廉,所以易于规模化生产。
技术实现要素:
针对现有技术存在的不足,本发明的目的在于提供一种3-环丙基环丁酮的制备方法,提供了一种新的可靠的技术路线和合成策略,在有机化学合成技术领域具有广阔的应用前景。
为实现上述目的,本发明提供了如下技术方案:一种3-环丙基环丁酮的制备方法,包括如下操作步骤:
步骤一、在金属催化剂的作用下,将环丙基乙炔溶于有机溶剂a,降温至0度之下,随后加入三氯乙酰氯,升至室温,反应7-9小时后经过滤干燥和减压浓缩后得到中间体;
步骤二、在还原催化剂和碱的作用下,步骤一中得到的中间体溶于有机溶剂b,并通入氢气,保持4-15atm下升温至50~60℃,反应15-18小时后过滤掉,减压浓缩以去除甲醇,然后减压蒸馏得到3-环丙基环丁酮。
通过采用上述技术方案,合成3-环丙基环丁酮的步骤分为两部,其原材料为环丙基乙炔和三氯乙酰氯,其中环丙基乙炔的结构式为:
进一步的,在步骤一中,所述有机溶剂a为乙醚;金属催化剂为zn-cu。
进一步的,在步骤一中,中间体的化学结构式为:
进一步的,在步骤一中,涉及的反应方程式为:
进一步的,在步骤一中,将过滤得到的滤液采用2-3mol/l的稀盐酸洗分层萃取一次,接着再采用2-3mol/l的氢氧化钠分层萃取一次,并采用无水硫酸钠干燥。
通过采用上述技术方案,在步骤一中,得到的中间体为黄色液体,对于产物为液体的净化处理,采用萃取的方式比较有效,第一次萃取可以去除产物中的氯化锌和氯化铜溶液,第二次萃取不仅可以中和第一次萃取后引入的氯离子,而且也不会影响中间体存在时的稳定性,随后采用无水硫酸钠进行吸水干燥,从而能够有效的去除产物中多余的水分,得到相对较纯的中间体。
进一步的,在步骤二中,所述还原催化剂为钯碳和/或氢氧化钯,所述有机溶剂b为甲醇。
进一步的,所述碱选自碳酸钠、碳酸氢钠、碳酸钾、三乙胺和二异丙基乙胺中的一种。
进一步的,在步骤一中,环丙基乙炔、zn-cu和三氯乙酰氯的重量比为1:(3~4):(5~7);每加入1g的环丙基乙炔,需要加入的26~32ml的乙醚。
通过采用上述技术方案,采用3倍以上的zn-cu此时能够吸收反应脱氯后的氯元素,有助于促进反应的正向进行,同时采用5倍以上的三氯乙酰氯,一方面也有利于反应的正向进行,另一方面能够保证环丙基乙炔原材料反应的充分性。
进一步的,在步骤二中,涉及的反应方程式为:
进一步的,在步骤二中,中间体、碳酸钠、无水钯碳的重量比为1:1:(0.3~0.5);每加入1g的中间体需要加入12~18ml的甲醇。
通过采用上述技术方案,优化试验后发现在上述配比范围内进行反应时得到的产物的产率较高。
进一步的,步骤一中的减压浓缩的真空度在0.05~0.08mpa,温度为20~30℃;而步骤二中的减压抽滤的真空度在0.06~0.08mpa,温度为30~40℃。
通过采用上述技术方案,在低温下减压抽滤掉甲醇,可以降低甲醇挥发或蒸发现象的发生,使减压抽滤能够稳定的进行。
进一步的,在步骤二中的减压蒸馏的条件为:在0.06~0.08mpa的真空度下,控制外温为60~80℃;内温为45~48℃。
通过采用上述技术方案,减压蒸馏是分离可提纯有机化合物的常用方法之一,因为液体的沸点是指它的蒸气压等于外界压力时的温度,因此液体的沸点是随外界压力的变化而变化的,如果借助于真空泵降低系统内压力,就可以降低液体的沸点,这便是减压蒸馏操作的理论依据。由此采用加压蒸馏降低液体的沸点后,不仅减低了提纯的难度、节能环保,而且加快了提纯的效率,十分实用。
综上所述,本发明具有以下有益效果:
1、本发明提供了一种新的可靠的技术路线和合成策略,在有机化学合成技术领域具有广阔的应用前景;
2、优化的,采用加压蒸馏降低液体的沸点后,不仅减低了提纯的难度、节能环保,而且加快了提纯的效率,十分实用。
附图说明
图1为一种3-环丙基环丁酮的制备方法中实施例1中步骤一的化学方程式;
图2为一种3-环丙基环丁酮的制备方法中实施例1中步骤二的化学方程式。
具体实施方式
以下结合附图对本发明作进一步详细说明。
实施例1:一种3-环丙基环丁酮的制备方法,包括如下操作步骤:
步骤一、将16.7g环丙基乙炔溶于500ml的乙醚,然后加入65.5g的zn-cu,随后温度降至0℃,将114g的三氯乙酰氯加入反应瓶中,然后升至室温,反应7小时后经过滤,得到的滤液采用2mol/l的稀盐酸洗分层萃取一次,接着再采用2mol/l的氢氧化钠分层萃取一次,并采用无水硫酸钠干燥,接着在真空度为0.05mpa,温度为30℃的条件下,减压浓缩后得到20g的中间体(黄色液体);
nmr:'h-nmr(cdcl3):δ1.43-1.49(m;4h),2.03-2.06(m;1h),6.06(s;1h),根据核磁共振氢谱的检查数据可知,上述中间体一的化学结构式为:
步骤二、将步骤一中得到的5g的中间体加入到高压反应釜中,然后加入5g的碳酸钠、2g的无水钯碳和75ml的甲醇,并通入氢气,并保持15atm下升温至50℃,反应15小时后过滤掉钯碳和碳酸钠固体,并在0.06mpa的真空度下,30℃的温度下减压抽滤以去除甲醇,在0.06mpa的真空度下,并控制溶液水浴外温为60℃;反应溶液的内温为45℃的条件下,减压蒸馏得到4.2g的3-环丙基环丁酮(无色溶液),其中3-环丙基环丁酮的产率为60.36%;
nmr:'h-nmr(cdcl3):δ0.15-0.66(m;4h),0.18-1.18(m;1h),1.98-2.38(m;1h),2.53-3.28(m,4h),根据核磁共振氢谱的检查数据可知,上述中步骤二中得到的产物的化学式为:
实施例2:一种3-环丙基环丁酮的制备方法,与实施例1的不同之处在于:原材料的用量、反应的温度、真空度和反应时间不同,其如下操作步骤:
步骤一、将16.7g环丙基乙炔溶于432.4ml的乙醚,然后加入50.1g的zn-cu,随后温度降至-3℃,将83.5g的三氯乙酰氯加入反应瓶中,然后升至室温,反应8小时后经过滤,得到的滤液采用2mol/l的稀盐酸洗分层萃取一次,接着再采用2mol/l的氢氧化钠分层萃取一次,并采用无水硫酸钠干燥,接着在真空度为0.08mpa,温度为20℃的条件下,减压浓缩后得到22g的中间体(黄色液体)。
实施例2中中间体的核磁共振氢谱数据如下:nmr:'h-nmr(cdcl3):δ1.43-1.49(m;4h),2.03-2.06(m;1h),6.06(s;1h).
步骤二、将步骤一中得到的5g的中间体加入到高压反应釜中,然后加入5g的碳酸钠、1.5g的无水钯碳和60ml的甲醇,并通入氢气,并保持15atm下升温至50℃,反应15小时后过滤掉钯碳和碳酸钠固体,并在0.08mpa的真空度下,40℃的温度下减压抽滤以去除甲醇,在0.08mpa的真空度下,并控制溶液水浴外温为80℃;反应溶液的内温为48℃的条件下,减压蒸馏得到4.1g的3-环丙基环丁酮(无色溶液),其中产率为64.82%。
实施例2中产物的核磁共振氢谱数据如下:nmr:'h-nmr(cdcl3):δ0.15-0.66(m;4h),0.18-1.18(m;1h),1.98-2.38(m;1h),2.53-3.28(m,4h).
实施例3:一种3-环丙基环丁酮的制备方法,与实施例1的不同之处在于:原材料的用量、反应的温度、真空度和反应时间不同,其如下操作步骤:
步骤一、将16.7g环丙基乙炔溶于534.4ml的乙醚,然后加入66.8g的zn-cu,随后温度降至-5℃,将116.9g的三氯乙酰氯加入反应瓶中,然后升至室温,反应9小时后经过滤,得到的滤液采用3mol/l的稀盐酸洗分层萃取一次,接着再采用3mol/l的氢氧化钠分层萃取一次,并采用无水硫酸钠干燥,接着在真空度为0.07mpa,温度为20℃的条件下,减压浓缩后得到18g的中间体(黄色液体)。
实施例3中中间体的核磁共振氢谱数据如下:nmr:'h-nmr(cdcl3):δ1.43-1.49(m;4h),2.03-2.06(m;1h),6.06(s;1h).
步骤二、将步骤一中得到的5g的中间体加入到高压反应釜中,然后加入5g的碳酸钠、2.5g的无水钯碳和90ml的甲醇,并通入氢气,并保持15atm下升温至50℃,反应15小时后过滤掉钯碳和碳酸钠固体,并在0.07mpa的真空度下,40℃的温度下减压抽滤以去除甲醇,在0.07mpa的真空度下,并控制溶液水浴外温为70℃;反应溶液的内温为46℃的条件下,减压蒸馏得到4.3g的3-环丙基环丁酮(无色溶液),其中产率为55.62%。
实施例3中产物的核磁共振氢谱数据如下:nmr:'h-nmr(cdcl3):δ0.15-0.66(m;4h),0.18-1.18(m;1h),1.98-2.38(m;1h),2.53-3.28(m,4h).
具体实施例仅仅是对本发明的解释,其并不是对本发明的限制,本领域技术人员在阅读完本说明书后可以根据需要对本实施例做出没有创造性贡献的修改,但只要在本发明的权利要求范围内都受到专利法的保护。
- 还没有人留言评论。精彩留言会获得点赞!