双氢青蒿素甾体缀合物及其制备方法和应用与流程
本发明涉及药物化学技术领域,具体涉及双氢青蒿素10-位羟基和各类甾体3-位羟基通过醚键缩合形成的缀合物及其衍生物的制备方法和应用。
背景技术:
青蒿素是从中药青蒿(artemisiaannual.)中分离、提取出的罕见的含有过氧基团的倍半萜内酯。青蒿素及其衍生物蒿甲醚、青蒿琥酯、双氢青蒿素等在临床上用于治疗疟疾,具有高效、速效、低毒的特点。除了它们的抗疟作用以外,在多年来的临床和实验室研究发现,青蒿素及其衍生物还具有优良的免疫抑制活性。
免疫抑制剂是一类具有免疫抑制作用的药物,主要功能为抑制机体异常的免疫反应,应用于自身免疫性疾病和过敏反应及器官移植后排斥反应的治疗。自身免疫性疾病是由于自身免疫应答过强或持续时间过长以至破坏自身正常组织结构并引起的相应临床症状;过敏反应即为机体抗原持续刺激或同一抗原再次刺激后产生的一种以生理功能紊乱和组织损伤为主要表现的病理性免疫反应;临床器官移植手术后,受者免疫系统可识别植物抗原并产生应答,移植物中免疫细胞也可以识别受者组织抗原并产生免疫应答,此为移植排异反应。这些症状都是机体“不适当”的免疫应答引起的,而免疫抑制剂在一定程度上能够起到治疗的作用,尤其是在器官移植排至反应的预防和治疗中起到了关键作用,在临床上得到了广泛的应用。
目前所有的免疫抑制剂可分为以下几种:
(1)抗代谢类:硫唑嘌呤(aza)、氨甲喋呤、霉酚酸酯(mmf)等;
(2)烃化剂:环磷酰胺等;
(3)皮质固醇类:强的松、地塞米松等;
(4)抗生素类:csa、fk506、雷帕霉素等;
(5)抗体类:抗淋巴细胞球蛋白(alg)、单克隆t淋巴细胞抗体(okt3)等;
(6)中草药:雷公藤多苷、冬虫夏草制剂等。
前三种为临床上已经使用的免疫抑制剂,这些药物的疗效虽然已经被证实,单只能改善症状,不能有效地从根本上控制疾病的发生和发展,且毒副作用大,不能长期持续应用。抗体类免疫抑制剂可以专一作用于淋巴细胞,但价格昂贵且有严重的不良反应,需要进一步改进。
随着近年来自身免疫性疾病的发病率不断增高,运用免疫抑制类药物进行治疗的需求增加。自身免疫性疾病大多为慢性或进行性疾病,需要长期用药,而现有免疫抑制剂的长期用药普遍观察到严重的副作用,因此临床上迫切需要新类型的高效低毒免疫抑制剂用于联合或交替用药。近年来在青蒿素类衍生物的免疫抑制活性研究中发现,经过结构改造合成的新型青蒿素类衍生物的免疫抑制活性有大幅提高,同时这类青蒿素衍生物保留了在体内低毒的特性,有望发展成为一类新型免疫抑制剂。
迄今为止,系统性红斑狼疮和类风湿性关节炎等自身免疫性疾病仍缺乏优良的治疗药物,这种状况已成为当今国际公认的医学难题。它们的发病机制及调控机理仍不十分清楚,这给治疗和免疫干预手段的设计带来了极大困难。目前国内外经典治疗方法包括使用激素和免疫抑制剂。激素治疗能暂时使症状缓解,但长期应用易诱发感染并产生诸多副作用,而且停药后易复发。虽然免疫抑制剂的出现和使用增加了临床对疾病进程的干预和治疗手段,但是除了价格昂贵外,长期使用还会产生较多毒副作用。因此,针对系统性红斑狼疮等自身免疫性疾病,研究和开发具有我国自主知识产权、高效低毒的新型免疫抑制剂已成为当今医学和药学研究的紧迫课题。
而干眼症是一种在临床上以不同程度结膜充血为特征的眼表炎症,尽管引起干眼的起始病因有所不同,但泪膜持续异常、眼表修复和防御功能损伤时,免疫相关的炎症反应成为干眼发病机制中最重要的环节。研究表明,泪膜长期异常引起泪液中抗炎成分(如乳铁蛋白)分泌减少,而眼表、泪腺等组织中的炎症细胞则产生炎症因子和蛋白酶,激活mapk信号通路,使泪液中炎症因子含量增多和蛋白酶活化,启动一系列炎症反应,引起t细胞在眼部的浸润【[j].investophthalmolvissci,2004,45:4293—4301】。因此,国内外均有尝试用免疫抑制剂治疗干眼症。
青蒿素及其衍生物在临床和实验室的研究中不断被证明具有一定的免疫抑制活性。为了研究和获得免疫抑制活性更高的化合物,药物工作者一直尝试在青蒿素母体结构上引入新的基团,合成了大量新的青蒿素类化合物。通过对这些新型青蒿素衍生物的研究,可以找到适用于临床的高效、低毒免疫抑制剂。中国科学院上海药物所研制的马来酸蒿乙醚胺(sm934),其水溶性、生物利用度与免疫抑制活性均显著高于双氢青蒿素,是近年来我国出现的较为优秀的免疫抑制类候选新药,且其结构简单,化学结构较为稳定,是近年来发现的一个针对系统性红斑狼疮的候选新药。而本发明以化合物4为代表的双氢青蒿素甾体缀合物经体外活性测试,免疫抑制活性远远优于sm934。在两个体外免疫抑制活性研究实验中:化合物对刀豆素a(cona)诱导的小鼠脾脏t淋巴细胞增殖的抑制实验及化合物对内毒素(lps)诱导正常小鼠脾脏b淋巴细胞增殖的抑制实验。化合物4的ic50值分别为0.015μm和0.015μm。而sm934对t淋巴细胞增殖抑制的ic50为1.2μm,化合物4的活性是其80倍;sm934对b淋巴细胞增殖抑制的ic50为2.6μm,化合物4的活性是其170倍。【internationalimmunopharmacology9(2009)1509–1517】
技术实现要素:
本发明要解决的技术问题是提供一种双氢青蒿素10-位羟基和甾体3-位羟基通过醚键缩合形成的缀合物及其衍生物,该双氢青蒿素甾体缀合物作为新的免疫抑制剂,其可以有效抑制免疫功能亢进或过度表达,单用或与其他药物联合用药,可用于治疗各类人体自身免疫性疾病。
为了解决上述技术问题,本发明通过如下技术方案实现:
本发明的一个方面,提供了一种具有通式(i)的双氢青蒿素10-位羟基和各类甾体3-位羟基通过醚键缩合形成的缀合物及其衍生物以及其异构体、或其可药用盐、或其前药分子,
其中,双氢青蒿素10-位羟基与各类甾体3-位羟基缩合而形成新的双氢青蒿素衍生物;
双氢青蒿素结构为(ⅱ):
双氢青蒿素10-位羟基可以为α或β构型;
其中,双氢青蒿素的10-位羟基和甾体化合物3-位羟基以醚键相连。双氢青蒿素10-位可以为α或β构型;甾体3-位可以是α或β构型;
通式(i)中,n=0~5;
通式(i)中,r1可以是6位或7位的h原子,或羟基,烷氧基或氨基及胺基,或酮羰基,或由6位和7位形成的双键;如果6位或7位是羟基或氨基及胺基,可以是α或β构型;
通式(i)中,r2可以是h原子或者羟基,烷氧基或氨基及胺基,或酮羰基;r2如果是羟基或氨基及胺基,可以是α或β构型;
通式(i)中,r3可以是氢,羧基及其衍生物,羟基及其衍生物,氨基及其衍生物,卤素,1-10个碳原子的烷基以及烯烃或炔烃,1-10个碳原子的醇或多元醇以及由醇羟基生成的衍生物,1-10个碳原子的羧酸或多元羧酸以及由羧基生成的衍生物,1-10个碳原子的烷基磺酸及其盐,1-10个碳原子的胺和由胺基生成的衍生物,1-10个碳原子的烷基亚胺和烷氧基亚胺,羟胺,羟胺磺酸酯及其盐,含有或不含杂原子的三元-八元的环烃或杂环烃,也包括r3与16位碳连接成环形成的衍生物。
本发明通式(i)中,双氢青蒿素的10-位羟基和甾体化合物3-位羟基缩合得到的缀合物的异构体包括其所有的异构体,如位置异构体,立体异构体和光学异构体。
优选的,所述通式(i)的衍生物包括下述具体结构的化合物:
在本发明中,双氢青蒿素的10-位羟基和甾体化合物3-位羟基缩合得到缀合物的可药用盐,包括锂盐,钾盐,钠盐,钙盐,镁盐,或者与天然及非天然的有机含氮化合物形成的有机盐。
在本发明中,双氢青蒿素的10-位羟基和甾体化合物3-位羟基缩合得到的缀合物前药分子,是指药物经化学结构修饰后得到的在体内能迅速转化为药效分子的化合物,设计前药分子的目的在于增加药物的生物利用度,加强靶向性,降低药物的毒副作用等。
在本发明的另一方面,双氢青蒿素的10-位羟基和甾体化合物3-位羟基缩合得到的缀合物还包括与辅料/载体形成的药物组合物,即含治疗有效量的上述双氢青蒿素-甾体缀合物或其异构体、或其可药用盐、或其前药分子,以及一种或多种药学上可接受的载体、稀释剂或赋形剂。
上述可接受的载体是无毒的、能辅助施用并且对结合物的治疗效果没有不利影响。此类载体可以是本领域的技术人员通常能得到的任何固体赋形剂、液体赋形剂、半固体赋形剂或者在气雾剂组合物中的气体赋形剂。固体药物赋形剂包括淀粉、纤维素、滑石、葡萄糖、乳糖、蔗糖、明胶、麦芽、稻米、面粉、白垩、硅胶、硬脂酸镁、硬脂酸钠、甘油硬脂酰酯、氯化钠、无水脱脂乳等。液体和半固体赋形剂可以选自甘油、丙二醇、水、乙醇和各种油,包括那些源于石油、动物、植物或人工合成的油,例如,花生油、豆油、矿物油、芝麻油等、优选的液体载体,特别是用于可注射溶液的,包括水、盐水、葡萄糖水溶液和甘醇。另外还可以在组合物中加入其它辅剂如香味剂、甜味剂等。
本发明的药物或药物组合物可以通过口服或其他给药方式给药,如注射,透皮给药,喷雾给药,直肠给药,阴道给药等。优选的给药方式是注射或口服,它可根据疾病程度调节。
本发明的药物组合物可以和其他免疫抑制剂联合用药,所述免疫抑制剂包括抗代谢类:硫唑嘌呤(aza)、氨甲喋呤、霉酚酸酯(mmf)等;烃化剂:环磷酰胺等;皮质固醇类:强的松、地塞米松等;抗生素类:csa、fk506、雷帕霉素等;抗体类:抗淋巴细胞球蛋白(alg)、单克隆t淋巴细胞抗体(okt3)等;中草药:雷公藤多苷、冬虫夏草制剂等。
本发明药物组合物的各种剂型可以按照药学领域的常规方法制备。例如使该化合物与一种或者多种载体混合,然后将其制成所需的剂型,如片剂、药丸、胶囊、半固体、粉末、缓释剂型、溶液、混悬液、配剂、气雾剂等。
在本发明的另一方面,还提供了上述双氢青蒿素的10-位羟基和甾体化合物3-位羟基在包括三氟化硼乙醚、酸性树脂、磷钨酸、樟脑磺酸等酸性试剂的催化下进行缩合反应得到缀合物的制备方法。
在本发明的另一方面,还提供了上述双氢青蒿素的10-位羟基和甾体化合物3-位羟基缩合得到的缀合物或其异构体、或其可药用盐、或其前药分子在制备治疗自身免疫性疾病的药物中的应用。
上述人体自身免疫性疾病包括红斑狼疮、类风湿关节炎、多发性硬化症、银屑病及干眼症等自身免疫性疾病等。
上述人体自身免疫性疾病也包括人体器官移植后的排斥反应。
1、本发明的衍生物,合成简单,化学稳定性好,毒性小,在体外试验中对t细胞增殖以及对干扰素ifn-γ都显示出非常强的抑制活性。
2、由于青蒿素类化合物的结构特点,其在体内半衰期只有40分钟左右,需要化合物尽快进入细胞而发挥作用。本发明的衍生物中,通过缀合的胆酸而增加了青蒿素对细胞的通透性和通透速度,使得化合物可以快速通过细胞膜表面的离子通道而进入细胞:因此其生物活性远远强于其他青蒿素衍生物,可以降低用量,进一步减少毒性。
3、本发明所得化合物免疫抑制活性比传统的青蒿素、双氢青蒿素、蒿甲醚、青蒿琥酯、β-氨基蒿乙醚马来酸盐(sm934)等更高,作为人体自身免疫性疾病治疗方面具有广阔的应用前景。
附图说明
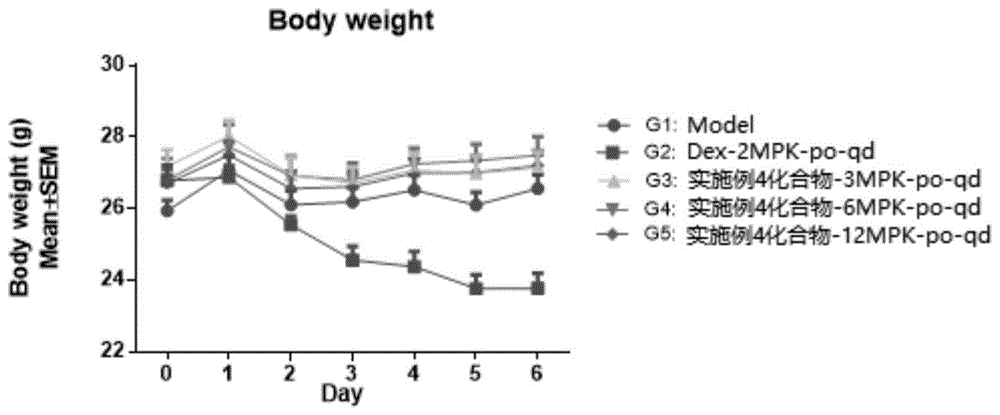
图1为体重变化图。
图2为右耳增加厚度变化图。
*表示g1和g2-5的统计学差异:****<0.0001,**<0.01。
图3为体重变化图。
图4为足垫厚度变化图。
*表示g1和g2-5的统计学差异:***<0.001,**<0.01,*<0.05。
具体实施方式
实施例1化合物1的制备:
-78℃下,三氟化硼乙醚(1ml)缓慢滴加至双氢青蒿素(569mg,2.0mmol)和胆酸(817mg,2.0mmol)的乙醚溶液(50ml)中,反应体系自然升至室温后搅拌过夜。薄层硅胶色谱监测至反应结束后,用饱和碳酸氢钠水溶液(30ml)缓慢淬灭反应,乙酸乙酯萃取(30ml×3),有机相合并后用水(50ml)洗涤1次,饱和食盐水(50ml)洗涤1次,无水硫酸镁干燥,过滤,滤液减压旋干即得粗产物。粗产物过硅胶柱纯化(石油醚:乙酸乙酯=1:1)得白色固体化合物726mg,产率:53.8%。
1hnmr(400mhz,cdcl3):δ5.47(s,1h),4.93(d,j=3.3hz,1h),4.00(s,1h),3.88(s,1h),3.51(t,j=11.0hz,1h),2.64–2.56(m,1h),2.47–2.21(m,4h),2.10–2.02(m,2h),1.99–1.85(m,5h),1.72(m,8h),1.53(m,5h),1.45(m,6h),1.40–1.30(m,5h),1.31–1.23(m,4h),1.16(m,2h),0.99(m,7h),0.89(m,8h),0.71(s,3h)。
13cnmr(100mhz,cdcl3):δ104.03(s),100.14(s),88.08(s),81.29(s),77.24(s),68.67(s),52.65(s),46.86(s),46.51(s),44.56(s),42.21(s),41.33(s),39.32(s),37.47(s),36.51(s),35.58(s),35.29(s),35.22(s),34.80(d),30.94–30.35(m),30.25(d),29.72(s),28.71(d),27.58(s),26.70(s),26.30(s),24.70(s),23.22(s),22.60(s),20.41(s),17.24(s),13.08(s),12.46(s)。
实施例2化合物2的制备
按照实施例1的类似方法制备得到白色固体。产率:54.8%。
1hnmr(400mhz,cdcl3):δ5.47(s,1h),4.91(d,j=3.4hz,1h),4.02(s,1h),3.67–3.55(m,1h),2.65–2.56(m,1h),2.48–2.25(m,3h),2.07–2.01(m,1h),1.92–1.81(m,5h),1.77–1.70(m,3h),1.68–1.58(m,4h),1.54(m,2h),1.50–1.34(m,12h),1.27(m,8h),1.18–1.06(m,2h),1.01(d,j=6.2hz,3h),0.96(d,j=6.3hz,3h),0.93–0.85(m,7h),0.71(s,3h)。
13cnmr(100mhz,cdcl3):δ179.27(s),104.03(s),100.49(s),88.10(s),81.30(s),77.24(s),73.32(s),52.62(s),48.36(s),47.33(s),46.46(s),44.55(s),42.00(s),37.43(s),36.51(s),36.06(s),35.26(s),35.04(s),34.66(s),34.30(s),33.79(s),32.64(s),31.05–30.54(m),29.86–29.32(m),28.98(s),27.35(d),26.29(s),26.06(s),24.64(d),23.62(s),23.34(s),20.38(s),17.33(s),13.20(s),12.77(s)。
实施例3化合物3的制备
按照实施例1的类似方法制备得到白色固体,产率:54.8%。
1hnmr(400mhz,cdcl3);δ5.46(s,1h),4.93(d,j=3.1hz,1h),3.88(s,1h),3.48(t,j=10.8hz,1h),2.65–2.55(m,1h),2.46–2.33(m,2h),2.27(m,1h),1.87(m,16h),1.52–1.13(m,21h),0.95(m,14h),0.66(s,3h)。
13cnmr(100mhz,cdcl3);δ179.74(s),104.02(s),100.40(s),88.09(s),81.30(s),77.49(s),77.25(s),68.70(s),55.79(s),52.63(s),50.53(s),44.59(s),42.73(s),41.33(s),39.56(s),39.33(s),37.47(s),36.51(s),35.86(s),35.30(d),34.80(d),32.88(s),30.86(d),29.12(s),28.18(s),26.29(s),24.63(d),23.70(s),22.90(s),20.64(s),20.41(s),18.27(s),13.20(s),11.78(s)。
实施例4化合物4的制备
按照实施例1的类似方法制备得到白色固体,产率:56.8%。
1hnmr(400mhz,cdcl3):δ5.46(s,1h),4.92(d,j=3.3hz,1h),3.61(m,2h),2.67–2.55(m,1h),2.47–2.34(m,2h),2.28(ddd,j=15.8,9.6,6.5hz,1h),2.04(m,2h),1.94–1.71(m,9h),1.71–1.55(m,4h),1.54–1.41(m,11h),1.40–1.21(m,9h),1.19–1.01(m,3h),0.98–0.89(m,12h),0.70(s,3h)。
13cnmr(100mhz,cdcl3):δ178.71(s),104.07(s),100.21(s),88.08(s),81.22(s),77.24(s),71.41(s),55.61(s),54.92(s),52.61(s),44.51(s),43.79(s),42.37(s),40.07(s),39.19(s),37.49(s),36.99(s),36.48(s),35.21(s),35.01(s),34.71(s),34.25(s),33.60(s),30.78(d),28.74(d),26.90(s),26.27(s),24.63(d),23.53(s),21.24(s),20.41(s),18.42(s),13.16(s),12.15(s)。
实施例5化合物5的制备
按照实施例1的类似方法制备得到白色固体,产率:65.5%.
1hnmr(400mhz,cdcl3):δ5.47(s,1h),4.92(d,j=3.3hz,1h),3.73–3.53(m,1h),2.66–2.56(m,1h),2.39(m,2h),2.27(ddd,j=15.8,9.7,6.4hz,1h),2.05(dt,j=14.5,3.7hz,1h),1.99–1.94(m,1h),1.93–1.70(m,8h),1.68–1.55(m,3h),1.51–1.21(m,21h),1.08(m,4h),1.00–0.89(m,13h),0.66(s,3h)。
13cnmr(100mhz,cdcl3)δ180.33(s),104.04(s),100.16(s),88.07(s),81.26(s),76.26(s),56.42(s),55.98(s),52.63(s),44.57(s),42.77(s),41.97(s),40.42(s),40.12(s),37.48(s),36.50(s),35.85(s),35.32(s),35.42(s),34.74(s),32.59(s),31.00(s),30.85(s),30.78(s),29.72(s),29.01(s),28.20(s),27.33(s),26.40(s),26.27(s),24.63(d),24.21(s),23.51(s),20.89(s),20.42(s),18.30(s),13.18(s),12.07(s)。
实施例6化合物6的制备
按照实施例1的类似方法制备得到白色固体,产率:49.5%。
1hnmr(400mhz,cdcl3);δ5.41(s,1h),4.96(d,j=3.5hz,1h),4.14–4.02(m,1h),3.64(dt,j=15.5,5.5hz,1h),2.68–2.58(m,1h),2.35(m,3h),2.12–1.95(m,2h),1.95–1.56(m,12h),1.56–1.21(m,16h),1.12(m,6h),1.02–0.89(m,10h),0.86(d,j=7.3hz,3h),0.65(s,3h)。
13cnmr(100mhz,cdcl3);δ179.13(s),104.09(s),98.78(s),88.20(s),81.19(s),77.24(s),72.49(s),71.73(s),56.21(s),55.93(s),52.64(s),47.88(s),44.45(s),42.88(s),39.99(d),37.55(s),36.51(s),36.00(s),35.39(d),34.60(d),31.64(s),30.24(s),30.36(s),30.44(s),30.75(s),30.77(s),30.87(s),29.67(s),28.11(s),26.26(s),24.64(d),24.22(s),23.77(s),20.75(s),20.41(s),18.25(s),13.16(s),12.02(s)。
实施例7化合物7的制备:
中间体7-1的制备:
氮气保护下,将8.0克去氧胆酸(20.38毫摩尔)和100毫升无水甲醇加入反应瓶中,然后再滴入2毫升浓硫酸。将反应液回流2小时,至薄层硅胶色谱显示反应完全为止。将反应液冷却到室温,减压浓缩,加入200毫升二氯甲烷后用5%的氢氧化钠水溶液和饱和食盐水洗涤。有机相经无水硫酸钠干燥、减压浓缩后真空干燥,得到8.20克白色固体,收率99%。
ms:[m+h]+=407.3
1hnmr(400mhz,cdcl3)δ:3.99(s,1h),3.67(s,3h),3.58-3.67(m,1h),2.33-2.43(m,1h),2.20-2.30(m,1h),1.50-1.92(m,13h),1.20-1.50(m,10h),1.03-1.18(m,1h),0.98(d,j=6.0hz,3h),0.92(s,3h),0.69(s,3h).
中间体7-2的制备:
氮气保护下,在三口圆底烧瓶中,将282毫克去氧胆酸甲酯7-1(0.69毫摩尔)和266毫克双氢青蒿素(0.94毫摩尔)溶于20毫升无水四氢呋喃中。溶液冷到-78℃后,滴入392毫克三氟化硼乙醚(2.76毫摩尔)。反应液缓慢升到室温后继续搅拌16小时。薄层硅胶色谱检测显示反应结束后,反应液用20毫升饱和碳酸氢钠水溶液淬灭并用20毫升乙酸乙酯萃取三次。合并有机相,用50毫升水和50毫升饱和食盐水洗涤,经无水硫酸钠干燥后减压浓缩。残余物经硅胶柱层析纯化(洗脱剂:石油醚/乙酸乙酯=10/1~3/1),得到150毫克白色固体,收率32.1%。
1hnmr(400mhz,cd3od)δ:5.45(s,1h),3.97(s,1h),3.65(s,3h),3.50-3.60(m,1h),2.47-2.57(m,1h),2.20-2.43(m,3h),2.00-2.09(m,1h),1.70-1.98(m,11h),1.04-1.70(m,26h),0.85-1.02(m,12h),0.70(s,3h).
化合物7的制备:
氮气保护下,在三口圆底烧瓶中,将30毫克硼氢化锂(1.115毫摩尔)悬浮在20毫升无水四氢呋喃中。0℃下,滴加150毫克中间体7-2(0.223毫摩尔)的5毫升无水四氢呋喃溶液。滴加完毕,反应液在室温下搅拌24小时。薄层硅胶色谱显示反应结束后,反应液用30毫升冷水淬灭并用30毫升乙酸乙酯萃取三次。合并有机相,用食盐水洗涤并经无水硫酸钠干燥,减压蒸干,残余物经硅胶柱层析纯化(洗脱剂:石油醚/乙酸乙酯=10/1~2/1),得到100毫克白色固体,收率69.5%。
mp:128.0-130.7℃
[α]22d:+94.8(c=0.1,ch3oh)
ms:[m-h+hcooh]-=689.3
1hnmr(400mhz,cdcl3)δ:5.46(s,1h),4.90(d,j=3.2hz,1h),4.02(s,1h),3.56-3.69(m,3h),2.55-2.65(m,1h),2.32-2.42(m,1h),2.00-2.08(m,1h),1.60-1.93(m,13h),1.30-1.60(m,19h),1.05-1.30(m,6h),1.02(d,j=6.4hz,3h),0.96(d,j=6.4hz,3h),0.85-0.95(m,6h),0.61(s,3h).
ir(kbr),cm-1:2936,2922,2863,1740,1726,1447,1374,1099,1009,981.
实施例8化合物8的制备
中间体8-1的制备:
室温下,将1.0克甘氨酸(13.32毫摩尔)溶于20毫升1,4-二氧六环和5毫升水的混合溶剂中,然后加入5.2克氯甲酸-9-芴基甲酯(20.1毫摩尔)和3.3克碳酸钾(23.88毫摩尔)。反应液在室温下搅拌12小时。滴加1摩尔/升的稀盐酸到反应液中,至ph值为1左右,然后用乙酸乙酯萃取三次。合并有机相,依次用水和饱和食盐水洗涤,经无水硫酸钠干燥,减压浓缩后真空干燥得到4.0克无色油状物。该粗品不经纯化,直接用于下步反应。
ms:[m+h]+=298.10
中间体8-2的制备:
氮气保护和室温下,向反应瓶中依次加入100毫克化合物7(0.155毫摩尔),55毫克中间体8-1(0.185毫摩尔),29毫克n,n'-二异丙基碳二亚胺(0.229毫摩尔),25毫克1-羟基苯并三唑(0.185毫摩尔),9毫克4-二甲氨基吡啶(0.074毫摩尔)和5毫升无水n,n-二甲基甲酰胺。反应液在室温下搅拌12小时。薄层硅胶色谱显示反应结束后,反应液用50毫升乙酸乙酯稀释,依次用水和饱和食盐水洗涤,无水硫酸钠干燥,减压浓缩。残余物经硅胶柱层析纯化(洗脱剂:乙酸乙酯/石油醚=1/4),得到110毫克白色固体,收率76.8%。
1hnmr(400mhz,cdcl3)δ:7.78(d,j=7.6hz,2h),7.62(d,j=7.6hz,2h),7.42(t,j=7.6hz,2h),7.32(d,j=7.6hz,2h),5.46(s,1h),5.31(m,1h),4.91(d,j=3.2hz,1h),4.42(d,j=7.2hz,2h),4.25(d,j=7.2hz,1h),4.10-4.20(m,2h),4.00(m,2h),3.56-3.66(m,1h),2.55-2.65(m,1h),2.32-2.42(m,1h),2.00-2.08(m,1h),1.00-1.95(m,38h),0.99(d,j=6.4hz,3h),0.96(d,j=6.4hz,3h),0.84-0.93(m,6h),0.68(s,3h).
化合物8的制备:
将50毫克中间体8-2(0.054毫摩尔)溶于5毫升乙腈中,室温下加入14毫克哌啶(0.16毫摩尔)并搅拌12小时。当薄层硅胶色谱显示反应结束后,反应液减压蒸干,残余物经硅胶柱层析纯化(洗脱剂:异丙醇/二氯甲烷=1/5),得到25毫克白色固体,收率65.9%。
mp:102.2-104.5℃
[α]18d:+97.8(c=0.1,ch3oh)
ms:[m+h]+=702.3
1hnmr(400mhz,cdcl3)δ:5.46(s,1h),4.90(d,j=3.2hz,1h),4.05-4.16(m,2h),4.02(s,1h),3.56-3.66(m,1h),3.46(s,2h),2.55-2.65(m,1h),2.32-2.42(m,1h),2.00-2.08(m,1h),1.30-1.94(m,32h),1.02-1.30(m,6h),0.99(d,j=6.4hz,3h),0.96(d,j=6.4hz,3h),0.85-0.94(m,6h),0.69(s,3h).
ir(kbr),cm-1:2936,2926,2863,1739,1447,1375,1194,1100,1010,965.
实施例9化合物9的制备
氮气保护下,向反应瓶中依次加入500毫克化合物7(0.775毫摩尔),103毫克烟酸(0.837毫摩尔),133毫克n,n'-二异丙基碳二亚胺(1.054毫摩尔),112毫克1-羟基苯并三唑(0.829毫摩尔),42毫克4-二甲氨基吡啶(0.344毫摩尔)以及10毫升无水n,n-二甲基甲酰胺。反应液在室温下搅拌12小时。薄层硅胶色谱显示反应完毕后,反应液用50毫升乙酸乙酯稀释,依次用水和食盐水洗涤,经无水硫酸钠干燥,减压浓缩。残余物经硅胶柱层析纯化(洗脱剂:乙酸乙酯/石油醚=1/3)得到450毫克白色固体,收率77.4%。
mp:98.0-101.7℃
[α]20d:+81.2(c=0.1,ch3oh)
ms:[m+h]+=750.5
1hnmr(400mhz,cdcl3)δ:9.24(s,1h),8.79(d,j=3.2hz,1h),8.30(m,1h),7.41(dd,j=3.2,7.6hz,1h),5.46(s,1h),4.90(d,j=3.6hz,1h),4.30-4.40(m,2h),4.03(d,j=2.8hz,1h),3.58-3.68(m,1h),2.56-2.65(m,1h),2.32-2.42(m,1h),2.00-2.08(m,1h),1.20-1.95(m,36h),1.06-1.30(m,6h),1.04(d,j=6.4hz,3h),0.96(d,j=6.4hz,3h),0.83-0.95(m,6h),0.70(s,3h).
ir(kbr),cm-1:2935,2923,2863,1725,1591,1447,1376,1281,1100,1021,1010,982.
实施例10化合物10的制备
中间体10-1的制备:
氮气保护下,室温下向反应瓶中依次加入400毫克化合物2(0.607毫摩尔),258毫克甘氨酸苄酯对甲苯磺酸盐(0.765毫摩尔),277毫克o-(7-氮杂苯并三唑-1-基)-n,n,n'-四甲基脲六氟磷酸盐(0.730毫摩尔)和10毫升无水n,n-二甲基甲酰胺。然后滴加226毫克n-乙基二异丙基胺(1.75毫摩尔)。反应液在室温下搅拌16小时。当薄层硅胶色谱显示反应完毕后,反应液中加入50毫升水,用乙酸乙酯萃取三次。合并有机相,用饱和食盐水洗涤三次,经无水硫酸钠干燥,减压浓缩。残余物经硅胶柱层析纯化(洗脱剂:乙酸乙酯/石油醚=20/1)得到401毫克白色固体,收率82.0%
1hnmr(400mhz,cdcl3)δ:7.32-7.42(m,5h),5.94(m,1h),5.46(s,1h),5.20(s,3h),4.90(d,j=3.2hz,1h),4.10(dd,j=2.0,5.2hz,2h),4.00(m,1h),3.76(t,j=6.0hz,1h),3.55-3.65(m,1h),2.56-2.65(m,1h),2.28-2.42(m,2h),2.10-2.21(m,1h),2.00-2.10(m,1h),1.70-1.92(m,13h),1.00-1.70(m,20h),1.00(d,j=6.0hz,3h),0.96(d,j=6.0hz,3h),0.82-0.93(m,6h),0.69(s,3h).
化合物10的制备:
将250毫克中间体10-1(0.31毫摩尔)溶于16毫升四氢呋喃和3毫升水的混合溶液中,0℃下加入60毫克氢氧化锂(2.50毫摩尔),反应温度逐渐升高到室温并搅拌4小时。当薄层硅胶色谱显示反应结束后,向反应液中滴加0.1摩尔/升的盐酸至ph值为6左右。用乙酸乙酯萃取三次。合并有机相,用饱和食盐水洗涤,经无水硫酸钠干燥后减压浓缩。残余物用硅胶柱层析进行纯化(洗脱剂:甲醇/二氯甲烷=1/100~5/100)得到147毫克白色固体,收率66.2%。
mp:142.5-145.2℃
[α]22d:+93.6(c=0.1,ch3oh)
ms:[m-h]-=714.5
1hnmr(400mhz,dmso-d6)δ:12.51(br,1h),8.01(t,j=5.2hz,1h),5.33(s,1h),4.79(d,j=3.2hz,1h),4.12-4.30(brs,1h),3.79(s,1h),3.67(d,j=5.2hz,2h),3.40-3.52(m,1h),2.30-2.41(m,1h),2.10-2.22(m,2h),1.95-2.09(m,2h),1.45-1.85(m,15h),1.05-1.42(m,17h),0.85-1.06(m,16h),0.61(s,3h).
ir(kbr),cm-1:3361,2936,2924,2864,1732,1656,1536,1448,1377,1194,1100,1021,1010,982.
实施例11化合物11的制备
中间体11-1的制备:
氮气保护下,向反应瓶中依次加入690毫克去氧胆酸(1.76毫摩尔),107毫克氯化铵(2.00毫摩尔),1135毫克2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(2.98毫摩尔),386毫克二异丙基乙基胺(2.98毫摩尔)和12毫升n,n-二甲基甲酰胺。反应液在室温下搅拌16小时。薄层硅胶色谱显示反应结束,反应液用乙酸乙酯稀释后用饱和食盐水洗涤三次,经无水硫酸钠干燥,减压浓缩,残余物经硅胶柱层析纯化(洗脱剂:乙酸乙酯/石油醚=1/5~2/1),得到679毫克白色固体,收率98.5%。
1hnmr(400mhz,cd3od)δ:3.97(m,1h),3.48-3.58(m,2h),2.22-2.32(m,1h),2.07-2.17(m,1h),1.73-1.96(m,7h),1.56-1.68(m,3h),1.06-1.56(m,13h),1.03(d,j=6.4hz,3h),0.94(s,3h),0.72(s,3h).
中间体11-2的制备:
氮气保护下,将含188毫克四氢铝锂(4.95毫摩尔)的15毫升无水四氢呋喃悬浮液冷到0℃,然后分批加入500毫克中间体11-1(1.28毫摩尔)。将反应液的温度逐渐升高到80℃并在该温度下回流2个半小时至薄层硅胶色谱显示原料消耗完毕。将反应液冷到0℃,小心滴入50毫升水,然后用100毫升乙酸乙酯萃取三次。合并有机相,用饱和食盐水洗涤,经无水硫酸钠干燥,减压浓缩。残余物经硅胶柱层析纯化(洗脱剂:二氯甲烷/甲醇=50/1~10/1)得到312毫克白色固体,收率64.6%。
1hnmr(400mhz,dmso-d6)δ:6.7-7.8(brs,2h),4.30-4.70(brs,1h),4.15-4.27(m,1h),3.78(s,1h),3.30-3.40(m,1h),3.16(s,1h),2.60-2.73(m,2h),1.70-1.86(m,4h),1.40-1.65(m,6h),1.20-1.40(m,10h),0.95-1.20(m,4h),0.96(d,j=6.4hz,3h),0.84(s,3h),0.61(s,3h).
中间体11-3的制备:
氮气保护下,室温下依次向反应瓶中加入100毫克中间体11-2(0.265毫摩尔),82毫克氯甲酸-9-芴基甲酯(0.317毫摩尔),5毫升二氯甲烷和51毫克三乙胺(0.504毫摩尔)。反应液在室温下搅拌16小时。薄层硅胶色谱显示反应结束后,反应液减压浓缩,残余物经c18硅胶柱层析纯化(洗脱剂:水/乙腈=30%-100%)得到95毫克白色固体,收率59.8%。
1hnmr(400mhz,cdcl3)δ:7.78(d,j=7.6hz,2h),7.61(d,j=7.6hz,2h),7.38-7.44(m,2h),7.30-7.37(m,2h),4.70-4.80(m,1h),4.41(d,j=6.8hz,2h),4.20-4.30(m,1h),4.00(m,1h),3.58-3.67(m,1h),3.06-3.74(m,2h),1.65-1.91(m,8h),1.50-1.60(m,5h),1.33-1.50(m,9h),1.00-1.25(m,4h),0.99(d,j=6.8hz,3h),0.95(s,3h),0.69(s,3h).
中间体11-4的制备:
氮气保护下,将683毫克中间体11-3(1.14毫摩尔)溶于60毫升无水四氢呋喃中并冷却到-78℃。1.00克三氟化硼乙醚(7.04毫摩尔)滴加到反应液中,随后反应液温度逐渐升高到室温。反应液在室温下搅拌16小时。当薄层硅胶色谱显示反应结束后,30毫升饱和碳酸氢钠滴加到反应液中,用乙酸乙酯萃取三次。合并有机相,依次用水和饱和食盐水洗涤,经无水硫酸钠干燥,减压浓缩。残余物经硅胶柱层析纯化(洗脱剂:石油醚/乙酸乙酯=20/1~3/1)得到613毫克白色固体,收率62.0%。
1hnmr(400mhz,cdcl3)δ:7.78(d,j=7.6hz,2h),7.61(d,j=7.6hz,2h),7.38-7.44(m,2h),7.30-7.37(m,2h),5.46(s,1h),4.91(d,j=3.6hz,1h),4.70-4.80(m,1h),4.41(d,j=6.8hz,2h),4.20-4.30(m,1h),4.00(m,1h),3.56-3.67(m,1h),3.10-3.25(m,2h),2.55-2.65(m,1h),2.32-2.42(m,1h),2.00-2.05(m,1h),1.79-1.94(m,5h),1.68-1.79(m,4h),1.22-1.68(m,22h),1.02-1.22(m,3h),1.11(d,j=6.4hz,3h),0.98(d,j=6.4hz,3h),0.85-0.95(m,6h),0.70(s,3h).
化合物11的制备:
将605毫克中间体11-4(0.698毫摩尔)溶于3毫升乙腈中,室温下加入178毫克哌啶(2.09毫摩尔)并在室温下搅拌16小时。薄层硅胶色谱显示原料消耗完毕。减压蒸干反应液,残余物溶于20毫升乙酸乙酯中,用饱和食盐水洗涤三次,经无水硫酸钠干燥,减压浓缩。残余物用c18硅胶柱层析纯化(洗脱剂:水/乙腈=30%-100%)得到340毫克白色固体,收率75.6%。
mp:151.7-154.1℃
[α]25d:+95.6(c=0.1,ch3oh)
ms:[m+h]+=644.4
1hnmr(400mhz,cdcl3)δ:5.45(s,1h),4.91(d,j=3.2hz,1h),4.01(s,1h),3.55-3.67(m,1h),2.74-2.90(m,2h),2.55-2.64(m,1h),2.00-2.09(m,1h),1.68-1.94(m,10h),1.48-1.68(m,9h),1.30-1.48(m,11h),1.03-1.30(m,7h),1.12(d,j=6.4hz,3h),0.99(d,j=6.4hz,3h),0.82-0.93(m,6h),0.69(s,3h).
ir(kbr),cm-1:2922,2863,1583,1447,1375,1343,1100,1010,982.
实施例12化合物12的制备
中间体12-1的制备:
氮气保护下,向反应瓶中依次加入500毫克去氧胆酸(1.27毫摩尔)、734毫克2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(1.93毫摩尔)、157毫克二甲胺盐酸盐(1.93毫摩尔)、20毫升无水n,n-二甲基甲酰胺和499毫克二异丙基乙基胺(3.86毫摩尔)。反应液在室温下搅拌过夜。当薄层硅胶色谱显示原料消耗完毕后,反应液用150毫升乙酸乙酯稀释,再用150毫升水洗涤4次。有机层用饱和食盐水洗涤后用无水硫酸钠干燥,减压浓缩得得451毫克白色粗品,直接用于下步反应,收率84.6%。
ms:[m+h]+=420.3
1hnmr(400mhz,dmso-d6)δ:4.45(d,j=4.0hz,1h),4.18(d,j=4.0hz,1h),3.78(m,1h),3.3-4.3(m,1h),2.94(s,3h),2.78(s,3h),2.23-2.43(m,1h),2.10-2.20(m,1h),1.40-1.85(m,10h),1.10-1.40(m,12h),0.8-1.08(m,8h),0.59(s,3h).
中间体12-2的制备:
氮气保护下,将156毫克氢化铝锂(4.11毫摩尔)的30毫升无水四氢呋喃悬浮液冷却到0℃,分批加入445毫克中间体12-1(1.06毫摩尔)。加毕,反应液逐渐升温到80℃并在该温度下反应2.5小时。薄层硅胶色谱显示反应结束后,将反应液冷却到0℃,滴加50毫升冷水到反应液中,然后用50毫升乙酸乙酯萃取三次。合并有机相,用食盐水洗涤并经无水硫酸钠干燥,减压浓缩,残余物经硅胶柱层析纯化(洗脱剂:二氯甲烷/甲醇=50/1~10/1)得到392毫克白色固体,收率91.2%。
ms:[m+h]+=406.3
1hnmr(400mhz,cdcl3)δ:3.99(m,1h),3.57-3.47(m,1h),2.38-2.47(m,2h),2.37(s,6h),1.03-1.90(m,26h),0.99(d,j=6.4hz,3h),0.92(s,3h),0.69(s,3h).
化合物12的制备:
氮气保护下,向反应瓶中依次加入380毫克中间体12-2(0.937毫摩尔)、865毫克双氢青蒿素(3.04毫摩尔)和60毫升无水四氢呋喃。将反应液冷却到-78℃后,滴加1.27克三氟化硼乙醚(8.95毫摩尔)。加毕,反应液温度逐渐升高到室温并搅拌16小时。薄层硅胶色谱显示反应结束后,反应液用冰水浴冷却到5℃,加入50毫升饱和碳酸氢钠水溶液。反应液用50毫升乙酸乙酯萃取3次。合并有机相,依次用水、饱和食盐水洗涤后经无水硫酸钠干燥,减压浓缩,残余物用硅胶柱层析纯化(洗脱剂:二氯甲烷/甲醇=100/1~40/1)得到182毫克类白色固体,收率28.9%。
mp:122.2-123.8℃
[α]17d:+83.8(c=0.1,ch3oh)
ms:[m+h]+=672.4
1hnmr(400mhz,cdcl3)δ:5.45(s,1h),4.90(d,j=3.2hz,1h),4.01(m,1h),3.55-3.66(m,1h),2.54-2.64(m,1h),2.30-2.45(m,6h),2.00-2.09(m,1h),1.57-1.92(m,13h),1.33-1.57(m,16h),1.02-1.32(m,12h),1.01(d,j=6.4hz,3h),0.95(d,j=6.4hz,3h),0.82-0.94(m,6h),0.69(s,3h).
ir(kbr),cm-1:2922,2861,2777,1462,1448,1375,1100,1020,1010,982.
实施例13化合物13的制备
中间体13-1的制备:
氮气保护下,在三口圆底烧瓶中,将6.0克胆酸甲酯(14.20毫摩尔)和5.48克双氢青蒿素(19.27毫摩尔)溶于300毫升无水四氢呋喃中。溶液冷到-78℃后,滴入8.05克三氟化硼乙醚(56.72毫摩尔)。反应液缓慢升到室温后继续搅拌16小时。薄层硅胶色谱检测显示反应结束后,反应液用300毫升饱和碳酸氢钠水溶液淬灭并用200毫升乙酸乙酯萃取三次。合并有机相,用300毫升水和300毫升饱和食盐水洗涤,经无水硫酸钠干燥后减压浓缩。残余物经硅胶柱层析纯化(洗脱剂:石油醚/乙酸乙酯=10/1~3/1),得到4.48克白色固体,收率45.79%。
1hnmr(400mhz,cdcl3)δ:5.45(s,1h),4.92(d,j=3.2hz,1h),3.99(s,1h),3.86(s,1h),3.75(t,j=6.4hz,3h),3.68(s,3h),3.42-3.53(m,1h),2.54-2.68(m,1h),2.19-2.45(m,4h),1.0-2.18(m,31h),0.95(m,6h),0.90(m,6h),0.70(s,3h).
化合物13的制备:
氮气保护下,在三口圆底烧瓶中,将708毫克硼氢化锂(31.51毫摩尔)悬浮在120毫升无水四氢呋喃中。0℃下,滴加4.48克中间体13-1(6.50毫摩尔)的30毫升无水四氢呋喃溶液。滴加完毕,反应液在室温下搅拌24小时。薄层硅胶色谱显示反应结束后,反应液用120毫升冷水淬灭并用100毫升乙酸乙酯萃取三次。合并有机相,用食盐水洗涤并经无水硫酸钠干燥,减压蒸干,残余物经硅胶柱层析纯化(洗脱剂:石油醚/乙酸乙酯=10/1~2/1),得到3.11克白色固体,收率72.3%。
mp:138.0-140.7℃
[α]22d:+91.2(c=0.1,ch3oh)
ms:[m-h+hcooh]-=705.2
1hnmr(400mhz,cdcl3)δ:5.45(s,1h),4.92(d,j=3.2hz,1h),4.01(s,1h),3.86(s,1h),3.63(t,j=6.0hz,2h),3.42-3.53(m,1h),2.54-2.64(m,1h),2.32-2.43(m,1h),2.18-2.28(m,1h),1.05-2.15(m,36h),1.02(d,j=6.4hz,3h),0.96(d,j=6.4hz,3h),0.89(m,6h),0.71(s,3h).
13cnmr(400mhz,cdcl3)δ:103.50,99.94,87.60,80.81,72.63,67.94,63.05,52.16,46.97,46.05,44.10,41.81,40.89,39.05,36.94,36.03,35.27,34.87,34.76,34.34,34.26,34.20,,31.29,30.42,28.88,28.60,27.97,27.06,26.43,25.80,24.20,24.13,22.65,22.23,19.88,17.23,12,66,12.08.
ir(kbr),cm-1:3419,2935,2922,2868,1447,1375,1099,1010,985.
实施例14化合物14的制备
中间体14-1的制备:
氮气保护下,依次向反应瓶中加入500毫克化合物13(0.756毫摩尔)、120毫克1-羟基苯并三唑(0.888毫摩尔)、45毫克4-二甲氨基吡啶(0.368毫摩尔)、143毫克n,n'-二异丙基碳二亚胺(1.133毫摩尔)、269毫克fmoc-甘氨酸(0.905毫摩尔)和10毫升无水n,n-二甲基甲酰胺。反应液在室温下搅拌12小时。薄层硅胶色谱显示反应结束后,反应液用100毫升乙酸乙酯稀释,再依次用饱和碳酸氢钠水溶液和饱和食盐水洗涤。有机相用无水硫酸钠干燥,减压浓缩,残余物经硅胶柱层析纯化(洗脱剂:乙酸乙酯/石油醚=1/2)得到550毫克白色固体,收率77.4%。
1hnmr(400mhz,cdcl3)δ:7.78(d,j=7.6hz,2h),7.61(d,j=7.6hz,2h),7.42(t,j=7.6hz,2h),7.33(t,j=7.6hz,2h),5.45(s,1h),5.31(m,1h),4,.91(d,j=3.6hz,1h),4.42(d,j=7.2hz,2h),4.26(t,j=7.2hz,1h),3.96-4.04(m,2h),3.78-3.90(m,3h),3.42-3.52(m,1h),2.55-2.64(m,1h),2.30-2.41(m,1h),2.13-2.24(m,1h),1.10-2.10(m,19h),1.00(d,j=6.4hz,3h),0.95(d,j=6.4hz,3h),0.85-0.95(m,6h),0.70(s,3h).
化合物14的制备:
将500毫克中间体14-1(0.532毫摩尔)溶解于45毫升乙腈中,加入137毫克哌啶(1.609毫摩尔)。反应液在室温下搅拌12小时至薄层硅胶色谱显示反应结束。将反应液减压浓缩,残余物经硅胶柱层析纯化(洗脱剂:甲醇/二氯甲烷=1/50)得到200毫克白色固体,收率52.4%。
mp:136.2-139.7℃
[α]22d:+77.4(c=0.1,ch3oh)
ms:[m+h]+=718.5
1hnmr(400mhz,dmso-d6)δ:5.334(s,1h),4.78(d,j=3.6hz,1h),4,15(d,j=3.6hz,1h),4.06(d,j=3.2hz,1h),3.94-4.04(m,2h),3.79(m,1h),3.61(m,1h),3.26(s,2h),2.30-2.40(s,1h),2.04-2.23(m,4h),1.90-2.03(m,2h),1.75-1.67(m,4h),1.50-1.76(m,8h),1.20-1.50(m,12h),0.95-1.20(m,4h),0.94(d,j=6.4hz,3h),0.89(d,j=6.4hz,3h),0.78-0.85(m,6h),0.59(s,3h).
ir(kbr),cm-1:3402,2921,2866,1738,1447,1375,1194,1099,1010,986.
实施例15化合物15的制备
氮气保护和室温下,向反应瓶中依次加入500毫克化合物13(0.756毫摩尔),100毫克烟酸(0.812毫摩尔),41毫克4-二甲氨基吡啶(0.336毫摩尔),130毫克n,n'-二异丙基碳二亚胺(1.030毫摩尔),137毫克1-羟基苯并三唑(1.014毫摩尔)以及10毫升无水n,n-二甲基甲酰胺。反应液在室温下搅拌12小时。当薄层硅胶色谱显示反应结束后,反应液用150毫升乙酸乙酯稀释,依次用150毫升水和150毫升饱和食盐水洗涤,经无水硫酸钠干燥,减压浓缩。残余物经硅胶柱层析纯化(洗脱剂:乙酸乙酯/石油醚=1/2)后得到400毫克白色固体,收率69.0%。
mp:124.9-126.8℃
[α]22d:+79.5(c=0.1,ch3oh)
ms:[m+h]+=766.5
1hnmr(400mhz,cdcl3)δ:9.24(s,1h),8.78(m,1h),8.30(d,j=8.0hz,1h),7.40(m,1h),5.44(s,1h),4.90(d,j=3.6hz,1h),4.33(m,2h),4.00(s,1h),3.85(s,1h),3.40-3.50(m,1h),2.52-2.63(m,1h),2.32-2.42(m,1h),2.14-2.29(m,1h),1.07-2.13(m,36h),1.03(d,j=6.8hz,3h),0.95(d,j=6.4hz,3h),0.87(m,6h),0.70(s,3h).
ir(kbr),cm-1:3419,2937,2925,2870,1724,1591,1375,1282,1099,1021,1011,986.
实施例16化合物16的制备
中间体16-1的制备:
氮气保护下,依次向反应瓶中加入400毫克化合物1(0.98毫摩尔)、151毫克甘胺酸甲酯盐酸盐(1.2毫摩尔),452毫克二异丙基乙基胺(3.5毫摩尔)、456毫克2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(1.2毫摩尔)和10毫升无水n,n-二甲酰基甲酰胺。反应液在室温下搅拌至薄层硅胶色谱显示反应完毕。用100毫升乙酸乙酯稀释反应液,然后依次用100毫升水和100毫升饱和食盐水洗涤。有机相用无水硫酸钠干燥,减压浓缩。残余物用硅胶柱层析纯化(洗脱剂:石油醚/乙酸乙酯=10/1~1/1)得到380毫克白色固体,收率52.0%。
1hnmr(400mhz,cdcl3)δ:6.18(t,j=5.2hz,1h),5.45(s,1h),4.92(d,j=3.2hz,1h),4.06(d,j=5.2hz,2h),3.98(m,1h),3,85(m,1h),3.77(s,3h),3.43-3.54(m,1h),2.55-2.64(m,1h),2.17-2.50(m,5h),1.10-2.17(m,h),0.93-1.00(m,6h),0.82-0.93(m,6h),0.70(s,3h).
化合物16的制备:
将120毫克中间体16-1(0.16毫摩尔)溶于5毫升四氢呋喃中,加入20毫克氢氧化锂(0.84毫摩尔)的1毫升水溶液。反应液在室温下搅拌4小时至薄层硅胶色谱显示反应结束。用1摩尔/升的硫酸氢钠水溶液调节反应液ph值为6左右。用80毫升乙酸乙酯萃取三次。合并有机相,用饱和食盐水洗涤,经无水硫酸钠干燥,减压浓缩。残余物经硅胶柱层析纯化(洗脱剂:二氯甲烷/甲醇=60/1~15/1)得到94毫克白色固体,收率80.3%。
mp:150.4-152.7℃
[α]22d:+75.2(c=0.1,ch3oh)
ms:[m-h]-=730.5
1hnmr(400mhz,dmso-d6)δ:7.81(brs,1h),5.32(s,1h),4.77(d,j=2.8hz,1h),4.10-4.20(br,1h),4.07(s,1h),3.78(s,1h),3.55-3.65(m,3h),2.30-2.40(m,1h),2.08-2.25(m,4h),1.90-2.08(m,3h),1.50-1.87(m,11h),1.10-1.50(m,16h),0.86-0.96(m,6h),0.80-0.86(m,6h),0.58(s,3h).
ir(kbr),cm-1:3399,2938,2922,2870,1634,1447,1376,1099,1020,1010,986.
实施例17化合物17的制备
中间体17-1的制备:
氮气保护下,依次向反应瓶中加入4.0克胆酸(9.79毫摩尔)、1.0克氯化铵(18.70毫摩尔)、3.8克o-(7-氮杂苯并三唑-1-基)-n,n,n′,n′-四甲基脲(10毫摩尔)、40毫升无水n,n-二甲基甲酰胺和2.5克二异丙基乙基胺(19.34毫摩尔)。反应液在室温下搅拌16小时。薄层硅胶色谱显示反应结束后,反应液用200毫升乙酸乙酯稀释,然后用100毫升水洗涤4次。有机相用饱和食盐水洗涤、经无水硫酸钠干燥,减压浓缩后真空干燥得到3.8克白色固体粗品、不经纯化直接用于下步反应,收率95.2%。
ms:[m+h]+=408.2
1hnmr(400mhz,dmso-d6)δ:7.20(s,1h),6.62(s,1h),3.90-4.20(m,3h),3.78(m,1h),3.61(m,1h),3.35(m,1h),3.13-3.23(m,1h),1.10-2.27(m,23h),0.92(d,j=6.4hz,3h),0.83(s,3h),0.59(s,3h).
中间体17-2的制备:
氮气保护下,将1.38克氢化铝锂(36.36毫摩尔)的60毫升无水四氢呋喃悬浮液冷却到0℃,将3.8克中间体17-1(9.32毫摩尔)分批加入。反应液温度逐渐升高到80℃,并在该温度下反应2.5小时。薄层硅胶色谱显示反应结束后,反应液冷却到0℃,缓慢滴加20毫升水,然后用100毫升乙酸乙酯萃取三次。合并有机相,用饱和食盐水洗涤后经无水硫酸钠干燥,减压浓缩后真空干燥,得到3.67克白色固体不经纯化直接用于下步反应,收率100%。
ms:[m+h]+=394.5
中间体17-3的制备:
氮气保护和室温下,将2.0克中间体17-2(5.08毫摩尔)溶于90毫升二氯甲烷中,再依次加入1.33克氯甲酸-9-芴基甲酯(5.14毫摩尔)和823毫克三乙胺(8.13毫摩尔)。反应液在室温下搅拌16小时。薄层硅胶色谱显示反应结束后,反应液减压浓缩,残余物经反相c-18硅胶柱层析纯化(洗脱剂用30%的乙腈水溶液),得到1.12克类白色固体,收率35.9%。
ms:[m+h]+=616.2
中间体17-4的制备:
氮气保护下,将890毫克中间体17-3(1.44毫摩尔)和570毫克双氢青蒿素(2.0毫摩尔)溶于50毫升无水四氢呋喃中。将反应液冷却到-78℃后,将838毫克三氟化硼乙醚(5.90毫摩尔)滴加到反应液中。反应液温度逐渐升高到室温并在室温下搅拌16小时。薄层硅胶色谱显示反应结束后,将反应液冷却到0℃,滴加30毫升饱和碳酸氢钠水溶液到反应液中,用乙酸乙酯萃取三次。合并有机相,用饱和食盐水洗涤,经无水硫酸钠干燥,减压浓缩,残余物用硅胶柱层析纯化(石油醚/乙酸乙酯=20/1~3/1)得到430毫克白色固体,收率33.8%。
ms:[m+nh4]+=899.4
化合物17的制备:
室温下,向430毫克中间体17-4(0.487毫摩尔)的20毫升乙腈溶液中加入120毫克哌啶(1.41毫摩尔)并在该温度下搅拌16小时。薄层硅胶色谱显示反应结束后,反应液中加入70毫升饱和碳酸钠水溶液,用70毫升乙酸乙酯萃取三次。合并有机相,用饱和食盐水洗涤,经无水硫酸钠干燥后减压浓缩。残余物用c18硅胶柱层析纯化(洗脱剂:水/乙腈=30%-100%)得到140毫克白色固体,收率43.6%。
mp:164.2-166.9℃
[α]24d:+87.8(c=0.1,ch3oh)
ms:[m+h]+=660.4
1hnmr(400mhz,dmso-d6)δ:5.70-6.40(br,2h),5.32(s,1h),4.78(d,j=3.2hz,1h),4.13-4.20(m,1h),4.07(d,j=3.2hz,1h),3.75-3.84(m,1h),3.58-3.66(m,1h),2.48-2.60(m,1h),2.30-2.40(m,1h),2.06-2.23(m,3h),1.90-2.03(m,2h),1.20-1.90(m,28h)m,0.95-1.20(m,4h),0.94(d,j=6.4hz,3h),0.91(d,j=6.4hz,3h),0.77-0.87(m,6h),0.60(s,3h).
ir(kbr),cm-1:3411,2935,2866,1448,1375,1100,1021,1011,986.
实施例18化合物18的制备
中间体18-1的制备:
氮气保护下,依次向反应瓶中加入1.2克胆酸(2.94毫摩尔),1.69克o-(7-氮杂苯并三唑-1-基)-n,n,n,n'-四甲基脲六氟磷酸盐(4.44毫摩尔),752毫克n-乙基二异丙基胺(5.82毫摩尔),46毫升无水n,n-二甲基甲酰胺以及2.2毫升2m的二甲胺无水四氢呋喃溶液(4.40毫摩尔)。反应液在室温下搅拌16小时,至薄层硅胶色谱显示反应结束。反应液浓缩蒸干后残余物溶于100毫升乙酸乙酯,依次用饱和氯化铵溶液、饱和碳酸氢钠水溶液和饱和食盐水洗涤,无水硫酸钠干燥,过滤浓缩,得到1.28克类白色固体直接用于下步反应,收率100%。
1hnmr(400mhz,dmso-d6)δ:4.32(d,j=4.4hz,1h),4.10(d,j=3.6hz,1h),4.01(d,j=3.2hz,1h),3.78(d,j=2.4hz,1h),3.60(s,1h),3.10-3.23(m,1h),2.95(s,3h),2.78(s,3h),2.10-2.33(m,4h),1.90-2.03(m,1h),1.55-1.85(m,6h),1.10-1.47(m,10h),0.94(d,j=6.4hz,3h),0.80(s,3h),0.59(s,3h).
中间体18-2的制备:
氮气保护下,向反应瓶中加入405毫克氢化铝锂(10.67毫摩尔)和60毫升无水四氢呋喃。分批加入1.28克中间体18-1(2.94毫摩尔)。当反应瓶中无气体产生后,将悬浮液缓慢升温到80℃并在该温度下回流2.5小时。当薄层硅胶色谱显示反应结束后,冷却反应液至0~5℃,分批加入十水硫酸钠,激烈搅拌。过滤,滤饼用150毫升乙酸乙酯洗涤。有机相用饱和食盐水洗涤,经无水硫酸钠干燥后减压浓缩,真空干燥,得到1.1克类白色固体直接用于下步反应,收率89%。
1hnmr(400mhz,cdcl3)δ:4.00(s,1h),3.86(s,1h),3.40-3.54(m,2h),1.05-2.35(m,31h),1.00(d,j=6.0hz,3h),0.90(s,3h),0.70(s,3h).
化合物18的制备:
氮气保护下,向反应瓶中依次加入1.1克中间体18-2(2.61毫摩尔),985毫克双氢青蒿素(3.46毫摩尔)和50毫升无水四氢呋喃。反应液降温到-78℃后,将1.45克三氟化硼乙醚(10.22毫摩尔)滴加到反应液中。滴毕,反应液缓慢升温到室温并继续搅拌16小时。薄层硅胶色谱显示反应结束后,20毫升饱和碳酸氢钠水溶液缓慢滴入反应液中,然后用乙酸乙酯萃取三次。合并有机相,用饱和食盐水洗涤,经无水硫酸钠干燥,减压浓缩。残余物经硅胶柱层析纯化(洗脱剂:二氯甲烷/甲醇=100/1~40/1),得到140毫克类白色固体,收率7.8%。
mp:118.5-121.3℃
[α]16d:+75.7(c=0.1,ch3oh)
ms:[m+h]+=688.4
1hnmr(400mhz,cdcl3)δ:5.46(s,1h),4.93(d,j=3.2hz,1h),3.96(s,1h),3.85(s,1h),3.40-3.57(m,1h),2.93-3.15(m,2h),2.85-2.95(m,6h),2.30-2.68(m,4h),1.10-2.10(m,35h),0.93-1.09(m,6h),0.80-0.93(m,6h),0.66(s,3h).
ir(kbr),cm-1:3412,2924,2868,1464,1446,1376,1098,1009,985.
实施例19化合物对cd3+cd28+抗体刺激的pbmc中t细胞的增殖抑制作用
实验原理:cd4+和cd8+是t淋巴细胞的两个重要的表面标志,cd4+t细胞能够辅助t淋巴细胞变为效应t细胞并且对b淋巴细胞转变为巨噬细胞和浆细胞有促进作用,而cd8+t细胞是一种细胞毒性t细胞,可分泌多种细胞因子,对机体的细胞免疫应答有着重要的作用,所以cd4+t和cd8+t细胞的数量是维持机体免疫功能平衡的关键。而自身免疫性疾病的发病机理主要是因过度免疫反应产生的大量免疫细胞,包括cd4+t和cd8+t细胞的大量产生,使得机体的免疫功能紊乱。因此通过检测药物对cd4+t和cd8+t细胞的增殖抑制活性便能初步评价其免疫抑制活性。
实验试剂和材料:
immunoculttmhumancd3/cd28tcellactivator:供应商stemcell;货号10971
easyseptmhumantcellisolationkit试剂盒:供应商stemcell;货号17951
pmbc(外周血单核细胞):供应商奥赛尔斯生物技术;货号pb004fc
celltraceviolet,供应商thermofisher,货号c34557
环孢素a(csa):供应商sigma-aldrich;货号239835
实验方法:用人总t细胞分离试剂盒(easyseptmhumantcellisolationkit,stemcell,negativeselection)从外周血单核细胞(pbmc)中分离t细胞,使用celltrace标记t细胞,加入抗cd3/cd28抗体刺激t细胞扩增,同时分别将化合物1-18及对照样品环孢素a(csa)与t细胞共同孵育120小时后,使用流式细胞仪分析cd4+t细胞与cd8+t细胞的增殖情况,同时设置celltrace标记的t细胞+抗cd3/cd28抗体(medium+cd3/cd28)和celltrace标记的t细胞不加抗cd3/cd28抗体两组作为系统对照,观察化合物1-18对cd4+t细胞与cd8+t细胞的增殖抑制活性。
实验步骤:
1.测试样品的准备
分别称取适量化合物1-18,加入dmso(0.1ml)溶解,以pbs定容到浓度为40mm,留作备用。
2.从外周血单核细胞(pbmc)中分离人总t细胞
按照人总t细胞分离试剂盒(easyseptmhumantcellisolationkit,stemcell)的说明书步骤,从pbmc中分离得到人总t细胞,实验步骤如下:
1)从液氮罐内取出冻存的pbmc,迅速置于37摄氏度水浴锅内,保证冻存管盖在水面上方,以防污染。用手不断摇动冻存管,使冻存的pbmc尽快融化。
2)使用75%酒精喷洒冻存管表面,并擦拭干净,转移到通风橱内。
3)将融化的pbmc转移至50ml离心管内。
4)缓慢加入10ml1640培养基(+10%热灭活牛血清+1%pbs)至50ml至离心管内并不断摇晃,使之混匀。
5)400g离心5min。
6)吸走上清,使用10ml1640培养基(+10%热灭活牛血清+1%pbs)垂悬细胞,400g离心5min,吸走上清液。
7)参考stemcelltotalhumantcell分离试剂盒,配制含有1%热灭活牛血清和1mmedta的分离液。
8)使用细胞计数仪对pbmc进行计数,使用分离液将pbmc垂悬至5x107cells/ml。
9)加入试剂盒中的enrichmentcocktail,50μl/ml。
10)混匀,室温孵育5min。
11)取出试剂盒中的rapidspherestmbeads,涡旋震荡30s。
12)将试剂盒内的rapidspherestmbeads加入样品内,40μl/ml。
13)混匀,室温孵育2min。
14)将混合物转移至流式管内。
15)将流式管插入磁极内,静止3min。
16)将上清倒出至新的离心管内,备用。
3.标记t细胞
根据产品说明书,用celltracetmviolet(celltracevioletcellproliferationkit,thermo)标记t细胞,实验步骤如下:
(1)将20ul的dmso与celltrace试剂混匀,混匀后浓度为5mm。
(2)配置标记buffer(dpbs+2%热灭活牛血清+2mmedta),使用10ml该buffer垂悬t细胞,400g离心5min。
(3)吸走上清,加入1ml该buffer,再加入1ulcelltracetmviolet,避光室温孵育20min。
(4)加入1640培养基(+10%热灭活牛血清+1%pbs),室温孵育5min。
(5)400g离心5min。
(6)吸走上清液,使用1640培养基(+10%热灭活牛血清+1%pbs)垂悬细胞,400g离心5min。
(7)使用1640培养基(+10%热灭活牛血清+1%pbs)垂悬celltrace标记好的t细胞,计数,并稀释至5x106cells/ml。
(8)向celltrace标记好的t细胞悬液里加入1/200体积的immunoculttmhumancd3/cd28tcellactivator,混匀。
(9)将细胞铺在96孔平底板内(corning,3599),每孔200ul。
(10)配置浓度为6.4mm,1.6mm,0.4mm,0.1mm,0.025mm的待测化合物(200x,终浓度分别为32μm,8μm,2μm,0.5μm,0.125μm,)每孔分别加入1ul待测化合物,混匀。
(11)同时设置未经celltrace标记的t细胞、celltrace标记的t细胞+immunoculttmhumancd3/cd28tcellactivator,celltrace标记的t细胞不加immunoculttmhumancd3/cd28tcellactivator作为三组系统对照。
(12)将96孔板置于细胞培养箱内孵育(37摄氏度,5%二氧化碳)。孵育5天。
(13)5天后,取出培养板,离心,吸取上清用于检测细胞因子ifnγ的分泌情况(humanifn-γelisakit)。
(14)使用facs检测t细胞的增殖情况。
4.facs检测t细胞增殖
(1)每孔加入200ulfacsbuffer(含有2%热灭活牛血清的dpbs)垂悬t细胞。
(2)400g离心5min。
(3)甩掉上清液,加入200ulfacsbuffer,400g离心5min,弃去上清液。
(4)每孔加入50ul染色抗体(1mlfacsbuffer内加入:1ulcellreactivefarredreactivedye,50μlbv605mouseanti-humancd4、50μlapc-cy7mouseanti-humancd8抗体),混匀,室温染色30min。
(5)每孔加入150μlfacsbuffer,400g离心5min,弃去上清。
(6)重复步骤(5),弃去上清。
(7)每孔加入100ulfacsbuffer垂悬细胞,上机检测。
统计方法:所有数据均以均数±标准差方式表示,所有各项指标检查结果经excel2000和graphpadprism5软件处理。
实验结果:
化合物1-18对cd4+t细胞增殖及对cd8+t细胞的增殖均具有较强抑制活性且呈一定的剂量依赖性,浓度越高,抑制作用越强。
实施例20化合物对t细胞分泌ifn-γ的抑制活性
实验原理:ifn-γ主要由th1细胞分泌,其生物学功能为诱导cd4+t细胞转变为th1细胞,促进t、b淋巴细胞的分化等。研究表明ifn-γ分泌水平的升高能够促进炎症的发生及机体组织的损伤,从而加重自身免疫性疾病的病情。故检测药物对ifn-γ的抑制活性能够评价其免疫抑制活性。我们采用ifn-γ酶联免疫吸附试验试剂盒(humanifn-γelisakit),通过酶联免疫吸附试验(elisa)检测化合物1-18分别作用于细胞后,在上清液中t细胞所分泌的ifn-γ的浓度,以此来观察各样品对ifn-γ的抑制活性。
实验试剂和材料:humanifn-γprecoatedelisakit试剂盒,供应商达科为,货号1110002
实验方法:将细胞上清液加入试剂盒提供的96孔板内,同时加入人ifnγ标准品的梯度稀释液及空白孔作为对照,室温孵育后,上清液内的ifnγ会结合到板底,洗板后加入抗人ifnγ的抗体(hrp标记),继续室温孵育,洗去未结合的抗ifnγ的抗体(hrp标记),加入底物室温显色15-20分钟,终止显色后读取od450数值。使用人ifnγ标准品的数据制作标准曲线,并计算各个待测样品内人ifnγ的浓度。
实验步骤:
1.将检测试剂盒内所有试剂置于室温平衡20分钟。
2.使用去离子水将试剂盒中50x洗液配置为1x洗液。
3.使用试剂盒中的稀释液将细胞上清液进行2.4倍稀释。
4.配置浓度分别为400pg/ml,200pg/ml,100pg/ml,50pg/ml,25pg/ml,12.5pg/ml的人ifnγ标准品。
5.将100μl标准品和100μl2.4倍稀释后的细胞上清液加入96孔板内。同时设置空白对照(加入100μl稀释液)
6.使用稀释液将试剂盒中biotinylatedantibody进行50倍稀释,每孔加入50μl,室温孵育2小时。
7.使用洗液洗板三次,300μl每孔。每次均需扣干孔内残留液体。
8.使用稀释液将试剂盒中streptavidin-hrp进行100倍稀释,每孔加入100μl,室温孵育20分钟。
9.重复步骤7,洗板三次。
10.加入试剂盒中的tmb(四甲基联苯胺)显色底物,每孔100μl,室温避光孵育15分钟。
11.每孔加入试剂盒中的100μl终止液,10分钟内使用读板仪读取od450值。
统计方法:所有数据均以均数±标准差方式表示,所有各项指标检查结果经excel2000和graphpadprism5软件处理。
实验结果:
使用酶联免疫吸附试验(elisa)检测了培养液中t细胞分泌的ifn-γ的浓度,发现化合物对t细胞分泌ifn-γ有较强的抑制作用,且呈一定的剂量依赖性,浓度越高,抑制作用越强。
实施例21化合物对刀豆素a诱导的小鼠脾脏t淋巴细胞增殖抑制实验
采用3h-胸腺嘧啶脱氧核苷(3h-thymidine)掺入法检测化合物1-18对cona(刀豆蛋白,concanavalina)诱导的正常小鼠脾脏淋巴细胞增殖功能的影响,评价样品的体外免疫抑制活性。
实验材料和试剂:雌性balb/c纯系小鼠,18-20克,来源:bk
刀豆蛋白a(concanavalina,cona):供应商sigma-aldrich,货号c7555
rpmi-1640培养基购自gibco;
3h-胸腺嘧啶脱氧核苷:武汉易泰科技有限公司上海分公司;
试验步骤:
1.小鼠脾脏淋巴细胞的制备:小鼠脱脊椎处死,无菌取其脾脏制备单个细胞悬液,红细胞裂解液去除红细胞,用含2%fbs的pbs洗3次后,用含10%fbs的rpmi-1640培养液洗1次并将细胞浓度调至4×106/ml。
2.3h-tdr掺入法检测实施例1-18化合物对小鼠脾脏淋巴细胞增殖功能的影响:小鼠脾脏淋巴细胞悬液100μl(4×105/孔)接种于96孔板,加入50μlcona(终浓度10μg/ml),不同浓度实施例1-18化合物50μl,测试终浓度为50、12.5、3.125、0.781、0.195、0.0488μm,每个浓度三复孔,总体积为200μl,并设相应的无cona对照孔以及无药物对照孔。37℃,5%co2培养箱中培养48小时。培养结束前8小时,每孔加入25μl3h-胸腺嘧啶脱氧核苷酸(10μci/ml)。继续培养至实验结束。将细胞用细胞收集仪收集至玻璃纤维膜上,加入闪烁液后于beta记数仪(microbetatrilux,perkinelmer)读取掺入细胞dna的3h-tdr量,以cpm值代表细胞增殖的情况。
统计方法:所有数据均以均数±标准差方式表示,所有各项指标检查结果经excel2000和spss11.0统计软件包处理。
实验结果:使用正常小鼠脾脏t淋巴细胞,在诱导剂cona的存在下,与系列化合物共孵育,检测化合物对小鼠脾脏t细胞增殖的抑制活性。结果显示化合物具有很强的抑制小鼠脾脏t淋巴细胞增殖的活性。
实施例22化合物对细菌脂多糖lps诱导正常小鼠脾脏b淋巴细胞增殖的抑制实验
3h-胸腺嘧啶脱氧核苷(3h-thymidine)掺入法检测化合物1-18对lps(lipopolysaccharid)诱导的正常小鼠脾脏b淋巴细胞增殖功能的影响,评价化合物的体外免疫抑制活性。
实验材料和试剂:雌性balb/c纯系小鼠,18-20克,供应商:bk
脂多糖lps(lipopolysaccharide):供应商j&k,货号l2762
rpmi-1640培养基:供应商gibco
3h-胸腺嘧啶脱氧核苷:供应商武汉易泰科技有限公司上海分公司
试验步骤:
1.小鼠脾脏淋巴细胞的制备:小鼠脱脊椎处死,无菌取其脾脏制备单细胞悬液,红细胞裂解液去除红细胞,用含2%fbs的pbs洗3次后,用含10%fbs的rpmi-1640培养液洗1次并将细胞调至5×106/ml。
2.3h-tdr掺入法检测化合物1-18对小鼠脾脏淋巴细胞增殖功能的影响:小鼠脾脏淋巴细胞悬液100μl(4×105/孔)接种于96孔板,加入50μllps(终浓度10μg/ml),不同浓度化合物1-1850μl,测试孔中化合物终浓度分别为50、12.5、3.125、0.781、0.195、0.0488μm,每个浓度三复孔,总体积为200μl,并设相应的无lps对照孔以及无药物对照孔。37℃,5%co2培养箱中培养48小时。培养结束前8小时,每孔加入25μl3h-胸腺嘧啶脱氧核苷酸(10μci/ml)。继续培养至实验结束。将细胞用细胞收集仪收集至玻璃纤维膜上,加入闪烁液后于beta记数仪(microbetatrilux,perkinelmer)读取掺入细胞dna的3h-tdr量,以cpm值代表细胞增殖的情况。
统计方法:所有数据均以均数±标准差方式表示,所有各项指标检查结果经excel2000和spss11.0统计软件包处理。
实验结果:在诱导剂的存在下,取正常小鼠脾脏b淋巴细胞与待测化合物共孵育,检测化合物1-18在不同浓度下对小鼠脾脏b淋巴细胞增殖的抑制活性。结果显示化合物1-18能够很强地抑制丝裂原细菌脂多糖(lps)诱导的正常小鼠脾脏b淋巴细胞增殖:
实施例23检测化合物对pha刺激人pbmc体外增殖抑制活性测试
检测化合物1-18对pha刺激人pbmc体外增殖的抑制活性。
实验试剂与材料:
sepmatetm-50:供应商stemcelltechnologies;货号86460
lymphoprep:供应商stemcelltechnologies;货号7861
dpbs:供应商biosera;货号lm-s2041/500
rpmi-1640:供应商gibco;货号11875-093
fbs(frenchorigin):供应商biosera;货号fb-1280/500
penicillin-streptomycin:供应商gibco;货号15140122
2-mercaptoethanol:供应商sigma-aldrich;货号m3148
pha:供应商sigma-aldrich;货号l8902-5mg
实验方法:分离人外周血单个核细胞(peripheralbloodmononuclearcell,pbmc),调整细胞浓度为1×106/ml,100μl每孔种入96孔板中。将实施例1-18化合物溶解至合适浓度的母液,以1:4梯度稀释,得到5个浓度梯度(2μm,0.5μm,0.125μm,0.031μm,0.008μm),加入到对应的孔中,每个浓度设置三复孔,阳性对照孔为2μm环孢菌素a(csa),阴性对照孔为rpmi-1640完全培养基(rpmi1640+10%fbs+1%penicillin-streptomycin+55μmβ巯基乙醇),同时每孔加入1μg/mlpha刺激pbmc细胞。培养箱中孵育72h后,每孔加入ctg,检测细胞的增殖情况。
实验步骤:
人外周血单个核细胞(pbmc)的分离
1)新鲜血液样本以相同体积的dpbs稀释,sepmate管中加入15mllymphoprep,将30ml稀释的血液样本缓慢的加在lymphoprep上面,注意不破坏界面。
2)加样完毕的sepmate管室温下1000xg离心25min。
3)收集包含pbmc的白细胞层至新的50ml离心管中,以40mldpbs洗两次,350xg离心5min。
4)弃上清,用rpmi-1640完全培养基重悬细胞,调整浓度为1×106/ml。
5)将pbmc细胞每孔100μl种入96孔板中,37℃,5%co2培养箱孵育15min。
6)用rpmi-1640完全培养液将待测化合物溶解至合适浓度的母液,以1:4梯度稀释,得到5个浓度梯度,每个浓度设置三复孔。
7)阳性对照组为csa,阴性对照组为rpmi-1640完全培养基。
8)将配制好的4倍浓度csa(体积为50μl/孔),4倍浓度待测化合物(体积为50μl/孔)加入到对应的孔中,同时加入1μg/mlpha(体积为50μl/孔)刺激pbmc细胞,每孔总体积为200μl。
9)将96孔板放入37℃,5%co2培养箱中培养72h。
10)72h后每孔加入ctg,检测细胞的增殖情况。
11)用graphpadprism6.0软件进行数据分析。
数据分析:以graphpadprism6.0软件分析数据。数据以平均值和标准误(standarderrorofthemean,sem)表示。
实验结果:
本次实验在pha刺激人pbmc体外增殖体系中,阳性对照2μmcsa可以抑制pbmc的增殖。化合物1-18均可以抑制pbmc的增殖,并呈现一定程度的剂量依赖性,ic50分别如下:
实施例24检测化合物4在dnfb(2,4-二硝基氟苯)诱导的小鼠耳肿胀模型上的药效学研究实验
实验材料和试剂:50只雌性icr小鼠,体重25-30克;来源:bk
dnfb(2,4-二硝基氟苯):sigma-aldrich(st.louis,mo,usa),cat:d1529。
丙酮:上海泰坦科技股份有限公司,cat:67-64-1。
橄榄油:上海凌峰化学试剂有限公司,cat:8001-25-0。
阳性对照化合物-dexamethasone(dex):damas-beta,纯度98%,cat:50-02-2。
实验方法:小鼠按体重随机分成5组,每组10只。分别为模型组、dex(2mpk,qd)组、化合物4高剂量(12mpk,qd)组、化合物4中剂量(6mpk,qd)组、化合物4低剂量(3mpk,qd)组。模型诱导的方法是:所有组别在实验的第0天和第一天腹部涂抹50μl的1%dnfb溶液,在第5天时右耳表面分别涂抹10μl的0.5%dnfb进行攻击,测量攻击前后的右耳厚度,利用差值对化合物的药效进行评价。
实验步骤:
1.配制1%dnfb,按丙酮:橄榄油比例为(4:1)配制混合液,即4ml丙酮内加入1ml橄榄油,混匀备用。50uldnfb加入混合液,混匀后分装成10管,每管0.5ml,封口,避光,每一笼小鼠使用一管。
2.致敏:第-1天所有动物麻醉剃毛,第0天和第1天,腹部涂抹50μl新鲜配制的1%dnfb(丙酮:橄榄油为4:1)。
3.第5天,所有动物测量右耳厚度数据,然后涂抹20μl的0.5%dnfb(10ul/边)。
4.第6天给药后再经过4小时,同时满足第5天右耳涂抹0.5%的dnfb经过了24h后(右耳涂抹0.5%dnfb24h后),用螺旋测微仪分别测每只小鼠右耳厚度,将右耳厚度减去第5天攻击前右耳厚度所得差值做为肿胀值。
5.给药:化合物(包括阳性药(dex))每天现配现用,口服,一天一次,连续给药7天。
6.体重:每天给药前称量。
统计分析:将各组收集数据用平均数和标准差表示(mean±sem)。对各种变化用graphpadprism软件进行分析。p<0.05认为有显著性差异,p<0.01认为有极显著差异。
实验结果:
1.体重变化
如图1所示,分组后,由于疾病造模的影响,实验初期所有组动物的体重都呈现缓慢下降的趋势,到实验第2天,所有动物(除了g2)的体重开始缓慢回升并保持平稳直到实验结束。g2即dex给药组的体重一直处于缓慢下降的趋势,推测是dex的副作用所致。实验显示化合物4的三个浓度都有较好的安全性。
右耳厚度
实验的第5天,所有动物麻醉后测量右耳的厚度,测量三次,取平均值。实验的第6天,所有动物麻醉后测量右耳的厚度,测量三次,取平均值。将攻击前后右耳的厚度差数据进行统计分析。攻击前后右耳的厚度差如图2所示,与模型组相比,dex治疗组(2mpk)显著降低了小鼠攻击前后右耳的厚度差(p<0.0001),而化合物4的低中高三个剂量的治疗组也都显著降低了小鼠攻击前后右耳的厚度差(p<0.01),且有一定的剂量依赖效应。结果提示,化合物4能够显著抑制dnfb诱导的耳肿胀,且药效与剂量有一定的相关性。
实施例25检测化合物4在绵羊红细胞引起的跖肿胀迟发超敏反应的药效学研究实验
实验材料和试剂:50只雌性icr小鼠;体重:25-30克;来源:bk
绵羊红细胞(srbc,浓度50%):货号:hq80073,批号:190617。
聚乙二醇400(peg400):alfa,货号:25322-68-3。
阳性对照化合物:醋酸泼尼松片(pns),每片含5mg的有效成分。
实验方法:小鼠按体重随机分成5组,每组10只;分别为模型组,pns(5mpk,qd)组,化合物4高剂量(6mpk,qd)组,化合物中剂量(2mpk,qd)组,化合物4低剂量(0.5mpk,qd)组。所有组别在实验的第一天腹腔注射绵羊红细胞,在第5天时再次左跖足垫皮下注射绵羊红细胞进行攻击,测量攻击后的左后跖足垫的厚度和右后跖足垫的厚度,利用差值对化合物的药效进行评价。
实验步骤:
1.在day1,g1-g5组所有动物腹腔注射2%(v/v)的srbc,每只注射0.2毫升(约1*108个srbc),进行动物免疫。
2.免疫后随即进行给药。
3.在day5,测量g1-g5组所有动物左后跖厚度,测量三次,取平均值;然后对g1-g5组所有动物测量部位皮下注射20%(v/v)的srbc,每只20ul(约1*108个srbc)进行攻击。
4.攻击后18-20小时给药,24小时测量(day6),所有动物麻醉后测量注射左后跖部位厚度以及右后跖部位厚度,各测量三次,取平均值。以攻击后左后跖足垫厚度和右足跖厚度差作为足肿胀度,分别予以数据分析。
5.给药:化合物每天现配现用;口服;一天一次,连续给药6天。
6.体重:每天给药前称量。
统计分析:将各组收集数据用平均数和标准差表示(mean±sem)。对各种变化用graphpadprism软件进行分析。p<0.05认为有显著性差异,p<0.01认为有极显著差异。
实验结果:
1.体重变化
如图3所示,分组后,由于疾病造模的影响,所有组动物的体重都呈现缓慢下降的趋势,其中以pns给药组的体重下降得最为明显。4个给药组分别与模型组相比,统计学上没有明显的差异(p>0.05)。
2.足垫厚度
实验的第6天,所有动物麻醉后测量左右后跖的厚度,测量三次,取平均值。将左后跖足垫厚度和右足跖厚度差数据进行统计分析。左右足跖的厚度差如图4所示,与模型组相比,化合物4的中高剂量治疗组也都显著降低了小鼠左右足跖的厚度差(p<0.01),有显著统计学差异。结果提示,化合物4能够显著抑制绵羊红细胞引起的跖肿胀迟发超敏反应,且药效与剂量有相关性。
以上所述实施例仅表达了本发明的实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!