量子点及其制备方法与流程
【技术领域】
本发明有关于一种量子点及量子点的制备方法,特别是有关于一种在较低的反应温度下制备量子点的方法。
背景技术:
量子点(quantumdots,qds)是纳米级的半导体材料,通常具有由约数百个原子到约数千个原子所形成的晶体结构。量子点具有光致发光(photoluminescence)特性。相较于现有技术已知的有机染料分子,量子点的优点包括:较高的荧光亮度、较佳的稳定性以及可调整的荧光波长等。
量子点是极具潜力的材料,且可应用于广泛的用途,例如,激光组件、光感测组件、内存组件、太阳光电组件、显示器组件、生物检测组件等。
然而,量子点的量产性低且价格昂贵,因此限制了其应用于各产业的潜力。再者,目前既有的量子点大多是包含镉的材料,对于环境的污染性高。基于环境友善的考虑,无镉的量子点逐渐成为研发的主流趋势。
因此,在本领域中仍需寻求更优选的量子点制备方法,以实现高效率生产无镉的量子点。
技术实现要素:
本发明的一实施例揭示一种量子点的制备方法,包括:(a)提供晶种组合物溶液,此晶种组合物溶液包括晶种组合物,且此晶种组合物为树状聚合物-金属纳米颗粒复合体;(b)在此晶种组合物溶液中加入核心半导体前驱物溶液,此核心半导体前驱物溶液包括第一半导体离子及第二半导体离子;以及(c)进行第一合成反应,以形成核心半导体材料包覆晶种组合物,此核心半导体材料是通过使第一半导体离子与第二半导体离子化合而形成的。
本发明的另一实施例揭示一种量子点,包括:晶种,此晶种包括金属纳米颗粒,且此金属纳米颗粒包括金、银、铂或钯;以及核心半导体材料,包覆晶种,其中核心半导体材料是通过使第一半导体离子与第二半导体离子化合而形成的。
为让本发明的上述和其他目的、特征、和优点能更明显易懂,下文特举出优选实施例,作详细说明如下:
【图式简单说明】
图1绘示根据本发明的一些实施例的晶种组合物的制备步骤的示意图。
图2绘示根据本发明的一些实施例的量子点的制备步骤的示意图。
图3绘示根据本发明的一些实施例的具有核-壳结构的量子点的制备步骤的示意图。
【符号说明】
102’~树状聚合物
102~晶种组合物
104~金属纳米颗粒
112~核心半导体前驱物
114~核心半导体材料
120~量子点
122~壳层半导体前驱物
124~壳层半导体材料
130~量子点
【具体实施方式】
在本说明书中,“约”、“大约”的用语通常表示在一给定值或范围的20%之内,优选是10%之内,且更优选是5%之内。在此给定的数量为大约的数量,意即在没有特定说明的情况下,仍可隐含“约”、“大约”的含义。
依据本发明一些实施例,提供一种量子点的制备方法,此制备方法使用树状聚合物-金属纳米颗粒复合体作为晶种组合物,进行量子点合成反应。由于此晶种组合物在反应溶液中的分散性与化学稳定性良好,且此晶种组合物与量子点前驱物之间的吸引力良好。使用此晶种组合物进行量子点的合成反应,可降低反应温度,且可改善量子点合成反应的制程窗口(processwindow)。因此,使用此制备方法能够高效率地生产量子点,进而降低生产量子点所耗费的时间与成本。
本发明的一些实施例提供一种量子点的制备方法,此方法包括以下步骤:
(a)提供晶种组合物溶液,其中此晶种组合物溶液包括晶种组合物,且此晶种组合物为树状聚合物(dendrimer)-金属纳米颗粒复合体;
(b)在此晶种组合物溶液中加入核心半导体前驱物溶液,其中此核心半导体前驱物溶液包括第一半导体离子及第二半导体离子;以及
(c)进行第一合成反应,以形成核心半导体材料包覆晶种组合物,其中此核心半导体材料是通过使第一半导体离子与第二半导体离子化合而形成。
以下配合图1详细说明上述量子点的制备方法的步骤(a)。
图1绘示根据本发明的一些实施例的晶种组合物102的制备步骤的示意图。请参照图1,首先,提供晶种组合物溶液。此晶种组合物溶液含有树状聚合物102’、金属盐化合物及溶剂。树状聚合物102’的结构绘示于图1的左侧部分。
金属盐溶解于溶剂中,而产生金属离子以及相对离子(抗衡离子,counterion)。其中,金属离子的电性与相对离子的电性相反。金属盐溶解所产生的金属离子受到树状聚合物102’中的官能基所吸引,而分散于树状聚合物102’中。另外,为了平衡电荷,相对离子也分散于树状聚合物102’中。金属离子以及相对离子吸附于树状聚合物102’的状态显示于图1的中间部分。
接着,对吸附有金属离子以及相对离子的树状聚合物102’提供适当的能量,使金属离子进行还原反应而形成金属原子。所产生的金属原子在树状聚合物102’中聚集而形成金属纳米颗粒104。因此,可得到含有晶种组合物102的晶种组合物溶液。晶种组合物102是一种树状聚合物-金属纳米颗粒复合体,且其结构绘示于图1的右侧部分。
提供适当的能量方法可包括但不限于:对晶种组合物溶液照射光线。由于不同的金属离子所需的还原反应能量不同,因此,可依据所使用的金属离子选择具有适当波长的光线。在一些实施例中,为了得到晶种组合物102,可使用波长200-350nm的紫外线照射晶种组合物溶液。在其他实施例中,可使用其他波长的光线照射晶种组合物溶液。
树状聚合物102’可作为用以合成金属纳米颗粒104的模板。如图1所示,树状聚合物102’具有控制良好的立体结构。树状聚合物102’的分子链末端上具有可吸引金属离子的官能基团,因此可有效地吸附金属离子。因此,可依据所欲合成的金属纳米颗粒104,选择合适的树状聚合物102’。举例而言,合适的树状聚合物102’包括但不限于:聚酰胺-胺(polyamidoamine,pamam)、聚丙烯胺-丙胺(polypropylenimine,ppi)。在一些实施例中,树状聚合物102’为聚酰胺-胺的树状聚合物。在这样的实施例中,树状聚合物102’中分子链末端的胺基可作为吸引金属离子的官能基团。
再者,为了控制金属纳米颗粒104的粒径大小与粒径均一性,可将树状聚合物102’的官能基团的摩尔数x1对金属纳米颗粒104的摩尔数x2的比率x1/x2调整为特定的范围内。
若比率x1/x2太低,则树状聚合物102’对金属离子的吸引力不够强,因而无法吸附足够量的金属离子。如此一来,可能导致金属纳米颗粒104的粒径太小而无法作为晶种,将不利于后续的量子点合成反应。反之,若比率x1/x2太高,则树状聚合物102’吸附的金属离子过多。如此一来,可能导致金属纳米颗粒104的粒径太大,或者是复数个金属纳米颗粒104被单一个树状聚合物102’包覆的情形。如此一来,作为晶种的金属纳米颗粒104的粒径过大或不均一,因而无法得到粒径均一的量子点。
在一些实施例中,树状聚合物102’的官能基团的摩尔数x1对金属纳米颗粒104的摩尔数x2的比率x1/x2为1-5。在另一些实施例中,x1/x2为1-4。在又一些实施例中,x1/x2为2-3。
此外,相较于内侧部分,树状聚合物102’的外侧部分的分子排列较为疏松。因此,树状聚合物102’的外侧部分具有可容纳金属纳米颗粒104的空间。此外,由于树状聚合物102’的立体障碍,可避免金属纳米颗粒104的粒径过大。由于不同代数的树状聚合物102’具有不同的立体障碍,因此,可通过选用合适代数的树状聚合物102’,而将金属纳米颗粒104的粒径控制在所需的范围内。
树状聚合物102’的代数越高,则树状聚合物102’中可用以形成金属纳米颗粒104的空间越小。因此,若树状聚合物102’的代数越高,则所形成的金属纳米颗粒104之粒径越小。再者,若树状聚合物102’的代数越高,则越容易形成单一个金属纳米颗粒104被单一个树状聚合物102’所包覆的状态。
若使用3代以下的树状聚合物102’,则所形成的金属纳米颗粒104之粒径可能会太大,进而造成后续合成的量子点的粒径过大。再者,若使用3代以下的树状聚合物102’,则有可能发生单一个金属纳米颗粒104被复数个树状聚合物102’包覆的情形,或者是复数个金属纳米颗粒104被单一个树状聚合物102’包覆的情形。如此一来,作为晶种的金属纳米颗粒104的粒径不均一,因而无法得到粒径均一的量子点。在一些实施例中,为了良好地形成所需的晶种组合物,可使用4.0代以上的树状聚合物102’。
树状聚合物102’的代数上限值,并无特别限制。然而,为了容易形成金属纳米颗粒104,并且降低成本,可使用8.0代以下的树状聚合物102’。在一些实施例中,可使用6.0代以下的树状聚合物102’。在另一些实施例中,可使用5.0代以下的树状聚合物102’。
金属盐化合物可作为金属纳米颗粒104的金属离子源。在本说明书中,所谓“金属离子”,包括仅由金属形成的阳离子、由金属与配位基所形成的阳离子、或由金属与配位基所形成的阴离子。
在后续的第一合成反应中,第一半导体离子与第二半导体离子会在金属纳米颗粒104的表面上进行反应,而形成核心半导体材料。若是金属纳米颗粒104的晶相(crystalphase)与核心半导体材料的晶相不相容,则难以在金属纳米颗粒104的表面形成核心半导体材料。换言之,核心半导体材料的合成反应的反应性不佳,因而导致生产效率的降低及生产成本的提高。此外,若是金属纳米颗粒104的晶相与核心半导体材料的晶相不兼容,则所产生的量子点的量子效率及稳定性皆不佳。在一些实施例中,为了提升核心半导体材料的合成反应的反应性,金属纳米颗粒104的晶相与核心半导体材料的晶相相容。再者,若是金属纳米颗粒104的晶相与核心半导体材料的晶相相同,则可更有利于核心半导体材料的形成,亦即,使用更为温和的反应条件(例如,较低的反应温度)进行核心半导体材料的合成反应。
举例而言,金属纳米颗粒104的晶相与核心半导体材料的晶相可各自独立地包括但不限于:立方晶相(cubic)、六方最密堆积晶相(hexagonal)。在一些实施例中,金属纳米颗粒104的晶相为立方晶相,且核心半导体材料的晶相亦为立方晶相。
金属纳米颗粒104的晶相可取决于金属纳米颗粒104的材料。在一些实施例中,金属纳米颗粒104可由至少一种金属材料所形成,且合适的金属材料可包括金、银、铂、钯或其他合适之金属。在另一些实施例中,金属纳米颗粒104可由单一金属材料所形成,且此金属材料可选自于金、银、铂及钯所组成的群组。在一些实施例中,金属纳米颗粒104可为金纳米颗粒。
可依据金属纳米颗粒104的材料,使用合适的金属盐化合物。在一些实施例中,金属盐化合物可包括但不限于:四氯金酸(chloroauricacid,haucl4)、硝酸银(silvernitrate,agno3)、四氯铂酸钾(potassiumtetrachloroplatinate,k2ptcl4)、四氯钯酸钠(disodiumtetrachloropalladate,na2pdcl4)。
此外,若金属纳米颗粒104的平均粒径太小,则无法作为晶种,因而无法降低合成量子点的反应温度,且无法改善合成反应的制程窗口。反之,若金属纳米颗粒104的平均粒径太大,则后续所合成的量子点的粒径也会太大,因而难以将量子点的粒径调整到所需的范围。
因此,为了合成所需的量子点,可将金属纳米颗粒104的平均粒径控制在特定的范围内。举例而言,金属纳米颗粒104的平均粒径可为小于或等于2nm。在一些实施例中,金属纳米颗粒104的平均粒径为0.5-2nm。在另一些实施例中,金属纳米颗粒104的平均粒径为0.8-1.6nm。在又一些实施例中,金属纳米颗粒104的平均粒径为1.0-1.4nm。
为了效率良好地形成晶种组合物102,可使用对树状聚合物102’及金属盐化合物的溶解性良好的溶剂。合适的溶剂可为极性溶剂。举例而言,晶种组合物溶液的溶剂可包括但不限于:水、甲醇、乙醇或上述的组合。在一些实施例中,晶种组合物溶液的溶剂为水。
以下配合图2详细说明上述量子点的制备方法的步骤(b)及(c)。
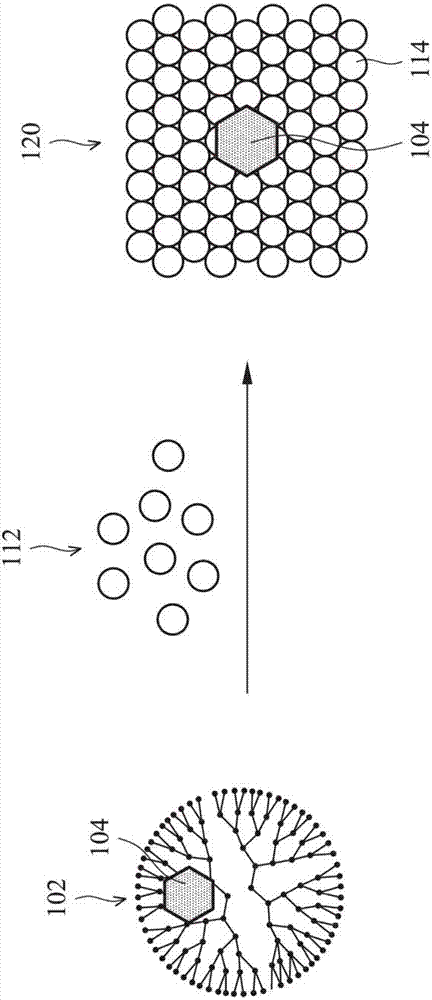
图2绘示根据本发明的一些实施例的量子点120的制备步骤的示意图。请参照图2,首先,提供包括晶种组合物102的晶种组合物溶液。如上所述,晶种组合物102是由树状聚合物102’包覆金属纳米颗粒104而形成。晶种组合物102的结构绘示于图2的左侧部分。
在上述步骤(b)中,在上述晶种组合物溶液中加入核心半导体前驱物溶液,此核心半导体前驱物溶液包括核心半导体前驱物112。为了简化说明,图2中仅绘示核心半导体前驱物112。实际上,核心半导体前驱物112可包括第一半导体离子的前驱物及第二半导体离子的前驱物。在接受适当的能量(例如,光能或热能)之后,第一半导体离子的前驱物可产生第一半导体离子,且第二半导体离子的前驱物可产生第二半导体离子。
在一些实施例中,第一半导体离子可为阳离子,且第二半导体离子可为阴离子。然而,这仅是为了举例说明,并非用以限定本发明。在另一些实施例中,第一半导体离子可为阴离子,且第二半导体离子可为阳离子。
在上述步骤(c)中,进行第一合成反应,以使第一半导体离子与第二半导体离子化合而形成核心半导体材料114,并且使核心半导体材料114包覆晶种组合物102而形成量子点120。量子点120的结构绘示于图2的右侧部分。
第一合成反应是在高温(例如,超过100℃)下进行。在进行第一合成反应的期间,晶种组合物102中的有机成分(即,树状聚合物102’)会因受热而分解。因此,在所形成的量子点120中,核心半导体材料114的内部并无树状聚合物102’的存在。换言之,量子点120是由核心半导体材料114包覆晶种(即,金属纳米颗粒104)所形成,如图2所示。
可依据所欲合成的量子点120,而使用合适的核心半导体前驱物112。用于形成量子点120的核心半导体材料114可包括ii/vi族化合物、iii/v族化合物或iv/vi族化合物。
在一些实施例中,第一半导体离子为阳离子,且可包括但不限于:锌离子、硼离子、铝离子、镓离子、铟离子、铊离子、硅离子或锗离子。在这样的实施例中,第一半导体离子的前驱物可为有机金属化合物、配位化合物、无机盐、其他合适的前驱物或上述的组合。举例而言,若第一半导体离子为铟离子,则第一半导体离子的前驱物可为氯化铟(incl3)。
在一些实施例中,第二半导体离子为阴离子,且可包括但不限于:氮离子、磷离子、砷离子、锑离子、硫离子、硒离子、碲离子或锡离子。在这样的实施例中,第二半导体离子的前驱物可为有机金属化合物、配位化合物、无机盐、其他合适的前驱物或上述的组合。举例而言,若第二半导体离子为磷离子,则第二半导体离子的前驱物可为三(二甲胺基)膦(p(n(ch3)2)3)。
更具体而言,第一合成反应是在高温下使核心半导体前驱物112产生第一半导体离子及第二半导体离子。接着,利用树状聚合物-金属纳米颗粒复合体(即,晶种组合物102)作为晶种,使第一半导体离子及第二半导体离子在金属纳米颗粒104的表面进行反应,而形成包覆金属纳米颗粒104的核心半导体材料114(即,量子点120)。在第一合成反应持续进行一段时间之后,通过降温或加入表面修饰剂,使量子点120停止成长。
依据本发明的一些实施例,利用树状聚合物-金属纳米颗粒复合体作为晶种。由于树状聚合物的官能基团对第一半导体离子(或第二半导体离子)具有吸引力,可将第一半导体离子聚集在金属纳米颗粒的附近。再者,树状聚合物的官能基团可以提高金属纳米颗粒与核心半导体前驱物之间的亲和力,进而降低异质成核的能障。因此,即使第一合成反应的反应温度较低,第一半导体离子及第二半导体离子也能够在金属纳米颗粒的表面进行反应,而形成量子点。换言之,本发明的晶种组合物能够明显地降低第一合成反应的反应温度。
在一些实施例中,第一合成反应的反应温度为170-190℃。在另一些实施例中,第一合成反应的反应温度为175-185℃。在又一些实施例中,第一合成反应的反应温度为180℃。
相较之下,若是不使用晶种而直接进行第一合成反应,或是使用不包括树状聚合物的晶种进行第一合成反应,则需要较高的反应温度(例如,约200℃或200℃以上)。在这样的情况下,当第一合成反应的反应温度为约170℃时,有可能无法行成量子点。
若是不使用晶种而直接进行第一合成反应,由于反应温度高(例如,200℃以上),第一合成反应的反应速度极快。换言之,在极短的时间(例如,小于60秒)之内,所有的量子点就会形成。在此情况下,难以控制量子点的粒径均一性。尤其是大量合成量子点时,很难在如此短的时间内使反应器的温度均一。换言之,在反应器的不同位置,温度不相同,且所合成的量子点的粒径也不相同。如此一来,量子点粒径不均一性的问题将变得更加严重。再者,由于量子点的反应时间太短(即,制程窗口太小),将导致可能进行的后续制程(例如,形成壳层)难以控制或操作。
另一方面,若量子点合成反应的制程窗口太大,则需要耗费过多的时间形成量子点。如此一来,将使量子点的生产效率明显降低。
相较之下,由于本发明的晶种组合物能够明显地降低第一合成反应的反应温度,因此,也适度降低了反应速度。换言之,通过使用本发明的晶种组合物,可将量子点合成反应的制程窗口控制在合适的范围内,进而实现生产效率与制程操作性的平衡。
在一些实施例中,第一合成反应的反应时间为90-300秒。在另一些实施例中,第一合成反应的反应时间为120-240秒。在又一些实施例中,第一合成反应的反应时间为150-180秒。
此外,由于树状聚合物表面具有官能基团,可提高金属纳米颗粒在溶剂中的分散性和稳定性,避免金属纳米颗粒聚集。因此,即使在合成之后保存一段时间(例如,数天),本发明的晶种组合物的晶种仍可具有反应活性。换言之,制程操作的灵活性很大。
相较之下,无机晶种的保存性差,合成之后很短时间内就会分解,而失去反应活性。因此,像这样的无机晶种必须在合成后立即使用,并且有很高比例的晶种可能会失去活性。制程操作的灵活性很小,经济效益也不佳。
此外,量子点具有光致发光特性。当量子点吸收能量较高的光后,其电子会从低能阶的状态被激发到高能阶的状态。之后,当被激发的电子从高能阶的状态回到低能阶的状态时,则会发射能量较低的荧光。
量子点所发射的荧光的波长会随着量子点的组成成分不同而有所不同。再者,对于由相同材料所形成的量子点,量子点的粒径越小,则能带隙(bandgap)越大。换言之,量子点所发射的荧光的波长也会随着量子点的粒径不同而有所不同。因此,通过控制量子点的组成成分及粒径,能够调整量子点的发光特性。
由于树状聚合物是控制良好的结构,因此,能够轻易控制金属纳米颗粒的粒径或晶相。因此,本发明的晶种组合物可有效地控制量子点的粒径,并实现良好的量子点粒径均一性。如此一来,可容易合成能够发射所需波长的光线的量子点,也能够形成发光波长范围较窄的量子点。
在形成上述量子点之后,也可视需要而对量子点实施其他制程。例如,可在核心半导体材料的表面再形成壳层,而形成具有核-壳(core-shell)结构的量子点。
本发明的另一些实施例提供一种具有核-壳结构的量子点的制备方法,此方法是在形成核心半导体材料之后,还包括以下步骤:
(d)在形成核心半导体材料之后,加入壳层半导体前驱物溶液,其中此壳层半导体前驱物溶液包括第三半导体离子及第四半导体离子;以及
(e)进行第二合成反应,以形成壳层半导体材料包复核心半导体材料,其中此壳层半导体材料是通过使第三半导体离子与第四半导体离子化合而形成。
以下配合图3详细说明上述量子点的制备方法的步骤(d)及(e)。
图3绘示根据本发明一些实施例的具有核-壳结构的量子点300的制备步骤的示意图。请参照图3,首先,提供量子点120。如上所述,量子点120是由核心半导体材料114包覆金属纳米颗粒104而形成。量子点120的结构绘示于图3的左侧部分。
在上述步骤(e)中,在形成核心半导体材料之后,加入壳层半导体前驱物溶液,此壳层半导体前驱物溶液包括壳层半导体前驱物122。为了简化说明,图3中仅绘示壳层半导体前驱物122。相似地,壳层半导体前驱物122可包括第三半导体离子的前驱物及第四半导体离子的前驱物。在接受适当的能量(例如,光能或热能)之后,第三半导体离子的前驱物可产生第三半导体离子,且第四半导体离子的前驱物可产生第四半导体离子。
在一些实施例中,第三半导体离子可为阳离子,且第四半导体离子可为阴离子。然而,这仅是为了举例说明,并非用以限定本发明。在另一些实施例中,第三半导体离子可为阴离子,且第四半导体离子可为阳离子。
在上述步骤(e)中,进行第二合成反应,以使第三半导体离子与第四半导体离子化合而形成壳层半导体材料124,并且使壳层半导体材料124包覆核心半导体材料114及晶种组合物102,而形成量子点130。量子点130的结构绘示于图2的右侧部分。
可通过选择壳层半导体材料124,而控制量子点130的能带隙,进而调整量子点130的发光特性。在一些实施例中,核心半导体材料114的能带隙宽于壳层半导体材料124的能带隙。在另一些实施例中,核心半导体材料114的能带隙窄于壳层半导体材料124的能带隙。
在第二合成反应中,若是壳层半导体材料124的晶相与核心半导体材料114的晶相不相容,则难以在核心半导体材料114的表面形成壳层半导体材料124。换言之,壳层半导体材料124的合成反应的反应性不佳,因而导致生产效率的降低及生产成本的提高。
在一些实施例中,为了提升第二合成反应的反应性,壳层半导体材料124的晶相与核心半导体材料114的晶相相容。举例而言,壳层半导体材料124的晶相与核心半导体材料114的晶相可各自独立地包括但不限于:立方晶相、六方最密堆积晶相。在一些实施例中,核心半导体材料114的晶相为立方晶相,且壳层半导体材料124的晶相亦为立方晶相。
可依据所欲合成的量子点130,而使用合适的壳层半导体前驱物122。用于形成量子点130的壳层半导体材料124可包括ii/vi族化合物、iii/v族化合物或iv/vi族化合物。为了控制量子点130的能带隙,壳层半导体材料124不同于核心半导体材料114。
在一些实施例中,第三半导体离子为阳离子,且可包括但不限于:锌离子、硼离子、铝离子、镓离子、铟离子、铊离子、硅离子或锗离子。在这样的实施例中,第一半导体离子的前驱物可为有机金属化合物、配位化合物、无机盐、其他合适的前驱物或上述的组合。举例而言,若第三半导体离子为锌离子,则第三半导体离子的前驱物可为硬酯酸锌(zn(c17h35coo)2)。
在一些实施例中,第四半导体离子为阴离子,且可包括但不限于:氮离子、磷离子、砷离子、锑离子、硫离子、硒离子、碲离子或锡离子。在这样的实施例中,第四半导体离子的前驱物可为有机金属化合物、配位化合物、无机盐、其他合适的前驱物或上述的组合。举例而言,若第四半导体离子为硫离子,则第二半导体离子的前驱物可为正十二硫醇(n-dodecylthiol,c12h26s)。
更具体而言,第二合成反应是在高温下使壳层半导体前驱物122产生第三半导体离子及第四半导体离子。接着使第三半导体离子及第四半导体离子在核心半导体材料114的表面进行反应,而形成具有核-壳结构的量子点130。在第二合成反应持续进行一段时间之后,通过降温或加入表面修饰剂,使量子点130停止成长。
由于晶种已被核心半导体材料包覆,因此,提升反应温度也不会使晶种分解。再者,提升反应温度可缩短第二合成反应的反应时间,改善量子点的生产效率。在一些实施例中,第二合成反应的反应温度为200-300℃。在一些实施例中,第二合成反应的反应时间为3-6小时。
本发明的另一些实施例提供一种量子点。请参照图2,量子点120包括晶种以及包覆晶种的核心半导体材料114。晶种包括金属纳米颗粒104,且金属纳米颗粒104包括金、银、铂或钯。核心半导体材料114是通过使第一半导体离子与第二半导体离子化合而形成的。
本发明的另一些实施例提供一种具有核-壳结构的量子点。请参照图3,量子点130包括晶种、包覆晶种的核心半导体材料114以及包复核心半导体材料114的壳层半导体材料124。壳层半导体材料124是通过使第三半导体离子与第四半导体离子化合而形成的。
量子点120及量子点130是通过上述方法所制备。如上所述,量子点120及量子点130具有粒径均一性良好、生产效率高及生产成本低等优点。
下文特举数个实施例,来说明本发明所述的量子点及制备量子点的方法。以下实施例中,所有化学药品皆购自sigma-aldrich。
在本说明书中以pamam树状聚合物-au纳米颗粒为例,说明树状聚合物-金属纳米颗粒复合体的制备方式。
【制备例1】pamam树状聚合物-au纳米颗粒复合体(i)的制备:
准备4.0代的聚酰胺-胺(pamam)树状聚合物(重均分子量为14215da,sigma-aldrich)的甲醇溶液,通过减压蒸馏将溶液中的甲醇去除,再将pamam树状聚合物溶解于水中,配制成浓度为6.24×10-6m的pamam水溶液。将四氯金酸(haucl4)溶解于水中,配制成浓度为0.2×10-3m的haucl4水溶液。将pamam树状聚合物的官能基团(即,胺基)的摩尔数设定为x1,且将haucl4的摩尔数设定为x2。以比率x1/x2等于4的方式,将pamam水溶液与haucl4水溶液混合。将混合后的溶液装入透明石英容器中,在暗箱中、室温下,以50w低压汞灯(光线波长:254nm)照射8小时。藉此制备含有pamam树状聚合物-au纳米颗粒复合体(i)的晶种组合物溶液。
【制备例2】pamam树状聚合物-au纳米颗粒复合体(ii)的制备:
将4.0代的pamam树状聚合物的甲醇溶液换为5.0代的pamam树状聚合物(重均分子量为28824da,sigma-aldrich)的甲醇溶液,除此之外,进行与制备例1相同的操作步骤,而制备含有pamam树状聚合物-au纳米颗粒复合体(ii)的晶种组合物溶液。
【au纳米颗粒的物性测定】
使用x-射线能量色散光谱仪(energydispersivex-rayspectroscopy,eds)分析制备例1及制备例2所得到的pamam树状聚合物-au纳米颗粒复合体。确认制备例1及制备例2所得到的纳米颗粒成分皆为金元素。
使用x射线衍射仪(x-raydiffractometer,xrd)分析制备例1及制备例2所得到的pamam树状聚合物-au纳米颗粒复合体。确认制备例1及制备例2所得到的au纳米颗粒的晶相皆为fcc立方结构,属于fm3m类晶相,晶格大小约0.2nm。
使用紫外光/可见光吸收光谱仪(ultraviolet/visibleabsorptionspectrometer)分析制备例1及制备例2所得到的pamam树状聚合物-au纳米颗粒复合体。确认制备例1所得到的au纳米颗粒在接近300nm及510nm处,分别有较强的吸收峰。确认制备例2所得到的au纳米颗粒在300nm处的uv吸收峰更加窄而且高。由此可证,相较于使用4.0代pamam树状聚合物所合成的au纳米颗粒,使用5.0代pamam树状聚合物所合成的au纳米颗粒,可提升粒径均一性。
使用透射电子显微镜(transmissionelectronmicroscopy,tem)分析制备例1及制备例2所得到的pamam树状聚合物-au纳米颗粒复合体。确认制备例2所得到的au纳米颗粒的粒径分布为2.0±0.3nm,平均粒径为2nm。不同粒径的au奈米颗粒有不同的uv吸收波峰位置。平均粒径系以tem(产品型号:jeoljem-2010f)影像量测计算,再辅以紫外线/可见光分光光谱仪(uv-visspectrophotometer,产品型号:lambda900)进一步确认。制备例2所得到的au纳米颗粒的粒径分布为1±0.2nm,平均粒径为1nm。
在本说明书中以inp/zns所形成的核/壳量子点为例,说明核/壳量子点的制备方式。
【实施例1】
在250ml三颈瓶中加入制备例1所得到的晶种组合物溶液10ml(溶液中pamam树状聚合物-au纳米颗粒复合体的重量为0.0013g)、氯化铟(incl3)1.2g及油酸(c18h34o2)23.02g,搅拌并使其均匀混合。将上述混合溶液在70℃脱氧(deoxygenation)3小时。接着,在氮气下将上述混合溶液迅速升温至190℃。在温度上升至190℃后,将三(二甲胺基)膦(p(n(ch3)2)3)2ml迅速注入上述混合溶液中。在注入三(二甲胺基)膦后,即开始进行第一合成反应。第一合成反应的反应时间为90秒。
将硬酯酸锌(zn(c17h35coo)2)3.794g、1-十八烯(ch3(ch2)15chch3,1-octadecene)20.1g及1-十二硫醇(c12h26s,1-dodecanthiol)12.144g混合并搅拌,而得到壳层半导体前驱物溶液。在第一合成反应结束之后,将上述壳层半导体前驱物溶液迅速注入上述三颈瓶中,并将溶液温度上升至250℃。使溶液温度在250℃下维持4.5小时,以进行第二合成反应。
在第二合成反应结束后,在溶液中加入过量的乙醇,以使核/壳量子点沉淀。接着,加入甲苯/乙醇混合溶剂,以6,000rpm离心纯化30分钟。最后,将所得到的inp/zns核/壳量子点分散于甲苯中。
【实施例2-3】
以表1的反应条件进行第一合成反应,除此之外,进行与实施例1相同的操作步骤,而制备inp/zns核/壳量子点。
【实施例4】
使用制备例2所得到的晶种组合物溶液10ml(溶液中pamam树状聚合物-au纳米颗粒复合体的重量为0.0013g)取代制备例1所得到的晶种组合物溶液,并且以表1的反应条件进行第一合成反应。除此之外,进行与实施例1相同的操作步骤,而制备inp/zns核/壳量子点。
【比较例1-5】
不加入晶种组合物溶液,并且以表1的反应条件进行第一合成反应,除此之外,进行与实施例1相同的操作步骤,而制备inp/zns核/壳量子点。
【inp/zns核/壳量子点的物性测定】
使用能量色散x-射线光谱仪分析实施例1-4及比较例1-5所得到inp/zns核/壳量子点。确认实施例1-4所得到的inp/zns核/壳量子点中皆含有金元素。确认比较例1-5所得到的inp/zns核/壳量子点中皆不含金元素。
使用光致发光荧光光谱仪(photoluminescencefluorescencespectrometer),以波长350nm的光源进行照射,分析实施例1-4及比较例1-5所得到的inp/zns核/壳量子点的光致发光荧光光谱。将inp/zns核/壳量子点的发光波长λ值及半峰全宽(fullwidthathalfmaximum,fwhm)值显示于表1中。发光波长λ值可用以判定量子点的粒径大小。若λ值越小,则代表量子点的粒径越小。半峰全宽值可用以判定量子点的粒径均一性。若半峰全宽值越小,则代表量子点的粒径均一性越佳。
表1
请参照表1的实施例1与比较例1,其中实施例1使用树状聚合物-金属纳米颗粒复合体作为晶种,而进行第一合成反应。所得到的inp/zns核/壳量子点具有相近的λ值,代表其粒径相近。实施例1与比较例1的反应时间相同。
然而,比较例1的反应温度为220℃,实施例1的反应温度仅为190℃。相似地,实施例2、3、4的反应温度分别低于比较例2、3、5。由此可证,本发明的晶种组合物能够明显地降低第一合成反应的反应温度。
再者,请参照表1的实施例1与比较例1,实施例1与比较例1的粒径相近。然而,比较例1的fwhm值为58nm,实施例1的fwhm值仅为52nm。相似地,实施例3、4的反应温度分别低于比较例4、5。由此可证,本发明的晶种组合物能够明显地提升量子点的粒径均一性。
此外,请参照表1的实施例3与比较例4,实施例3与比较例4的反应条件相同。然而,实施例3的λ值为512nm,比较例4的λ值仅为407nm。代表在相同的低温(170℃)反应条件下,实施例3的量子点粒径大于比较例4的量子点粒径。由此可证,本发明的晶种组合物能够明显地改善第一合成反应的反应性。
综上所述,本发明提供一种量子点的制备方法。此制备方法使用树状聚合物-金属纳米颗粒复合体作为晶种组合物,进行量子点合成反应。本发明的量子点的制备方法至少具备以下优点:
(1)此晶种组合物的分散性与化学稳定性良好,且此晶种组合物与量子点前驱物之间的吸引力良好。因此,使用此晶种组合物,可降低量子点合成反应的反应温度。
(2)此晶种组合物能够降低量子点合成反应的反应温度及反应速度。因此,使用此晶种组合物,可将量子点合成反应的制程窗口控制在合适的范围内,可改善制程操作性。
(3)此晶种组合物的保存性佳,因此,在合成之后可以保存一段时间,可改善制程操作的灵活性。
(4)使用此晶种组合物,能够高效率且低成本地生产量子点。
(5)此量子点的制备方法中不使用含镉或汞的原料,制程及产品的污染性低。
虽然本发明已以数个优选实施例揭露如上,然其并非用以限定本发明,任何所属技术领域中的技术人员在不脱离本发明的精神和范围内,应可作任意的更动与润饰,因此本发明的保护范围应视所附权利要求所界定的范围为准。
- 还没有人留言评论。精彩留言会获得点赞!