一种由铜基催化剂直接加氢乙酰丙酸制备1,4‑戊二醇的方法与流程
本发明属于化工技术领域,涉及一种面向1,4-戊二醇合成的新型铜基加氢反应催化剂的组成及制备方法,和采用该催化剂由乙酰丙酸直接合成1,4-戊二醇的制备方法。
背景技术:
作为人类生存和发展的重要物质基础,化石资源支撑了19世纪到20世纪近200年来人类文明的进步和经济社会发展。它们不仅是人类消耗的最主要能源,也是满足人类日常所需的各种化学品的原料。然而,化石资源的不可再生性和人类对其的巨大消耗,使化石能源正在逐渐走向枯竭。为了解决能源危机,众多的科研工作人员致力于开发太阳能、风能、核能等新能源,但无法解决资源性问题。而通过开发利用生物质,既可以用生物质能解决能源危机,也可以以生物质资源代替石油等石化资源,作为各种化工品的基础原料。
1,4-戊二醇是一种潜在的聚酯类高分子单体,与二酸反应可以制得聚酯材料,因而需求量巨大。目前,1,4-戊二醇的生产还比较困难,可行的生产路线主要由乙酰丙酸,乙酰丙酸酯和γ-戊内酯催化加氢而来。乙酰丙酸酯与γ-戊内酯主要是由生物质水解产物乙酰丙酸生产而来,分离的生产工序增加了1,4-戊二醇的制造成本。直接由乙酰丙酸催化加氢生产1,4-戊二醇,反应条件极为苛刻,目前只能采用贵金属催化剂,反应条件的限制与贵金属催化剂昂贵的成本,制约了1,4-戊二醇的大规模生产。然而,至今为止,高效可回收且廉价的非贵金属催化剂用于1,4-戊二醇的生产国内外还未见报道。因此开发新型且经济高效的催化剂用于由乙酰丙酸直接生产1,4-戊二醇十分重要。
技术实现要素:
本发明的目的是提供一种由铜基催化剂直接加氢乙酰丙酸制备1,4-戊二醇的方法,包括一种加氢制备1,4-戊二醇的铜基催化剂的方法,以及在此催化剂上可实现的由乙酰丙酸直接加氢高效制备1,4-戊二醇的新方法。
本发明所述的一种加氢制备1,4-戊二醇的铜基催化剂的方法,其特征在于其主体活性组分为铜,以铁及硼为助催化组分,催化剂中三种元素的重量百分比为:铜含量为50%-99%,铁含量为0.9%-40%,硼含量为0.1%-10%。此类铜基催化剂,均采用液相化学还原法制备,具体制备步骤如下:配制包含铜及铁的总金属浓度为0.01-2mol/l的盐溶液a,通入氮气保护,放置在冰水浴中,机械剧烈搅拌;将配制好的还原剂混合液b用蠕动泵滴加至盐溶液中,滴加时间为1-2h;滴加完毕后继续反应2h以等待充分还原;将反应后的产物进行抽滤,用去离子水抽滤5次,无水乙醇抽滤3次;将得到的催化剂保存在无水乙醇中。
所述的盐溶液a,其总金属盐溶液浓度优选为0.1mol/l;其铁占总金属摩尔比例控制在50%以内。
所述的盐溶液溶剂为水、乙醇、甲醇或乙二醇,优选为水;金属盐为硝酸盐或氯化盐,优选为硝酸盐。
所述的还原剂混合液b,其还原剂为硼氢化钠或者硼氢化钾,优选为硼氢化钠;混合液b中还原剂浓度为2mol/l,溶剂为水;还原剂总量与盐溶液a中金属的摩尔比控制为4﹕1;在混合液b中加入氢氧化钠溶液调节ph值为12-14,所用氢氧化钠溶液浓度为0.2mol/l。
本发明所述的一种由铜基催化剂直接加氢乙酰丙酸制备1,4-戊二醇的方法,包括以下步骤:在上述条件所制铜基催化剂存在的条件下,将反应溶剂和反应物乙酰丙酸直接混合,通入氢气进行一锅式加氢反应,反应完毕后得到1,4-戊二醇。
所述的反应溶剂包括水、甲醇、乙醇、十二烷、乙醚或1,4-二氧六环中一种或两种以上,优选1,4-二氧六环。制备条件:乙酰丙酸的浓度控制为0.1-0.5mol/l,反应温度为60-250℃,氢气压力为0.1-6mpa,反应时间是0.1-24h。
本发明的技术效果:与现有技术相比,制备出了廉价的非贵金属新型铜基催化剂,其合成方法简单,成本低廉,无毒无害,便于规模化生产,同时具有磁性,易于使用后回收及重复使用;以该催化剂可高效实现由乙酰丙酸直接加氢转化为1,4戊二醇,选择性好,1,4-戊二醇的总产率可高达85%,表明该合成工艺方法具有较高的潜在经济价值。
附图说明
图1为本发明催化剂的x射线衍射(xrd)图谱。
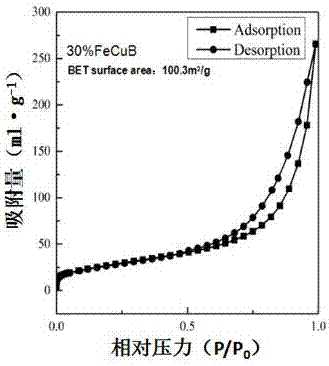
图2为本发明催化剂的氮气脱附吸附(bet)图谱。
图3为本发明催化剂的透射电子显微镜(tem)图,图(a)为总体形貌,图(b)为颗粒形貌。
具体实施方式
根据下述实施例,可以更好地理解本发明。实施例所描述的内容仅用于说明本发明,而不应当也不会限制权利要求书中所详细描述的本发明。
实施例1。
催化剂30%fecub的制备:30%代表铁占总金属(铁+铜)摩尔比例为30%,配制包含九水硝酸铁1.01g,三水硝酸铜5.45g与水250ml的盐溶液a。通入氮气保护,放置在冰水浴中,机械剧烈搅拌。配制包含硼氢化钠3.79g,氢氧化钠0.4g与水50ml的还原剂混合液b。将配制好的还原剂混合液b用蠕动泵滴加至盐溶液a中,滴加时间为1.5h.滴加完毕后继续反应2h以等待充分还原。将反应后的产物进行抽滤,用去离子水抽滤5次,无水乙醇抽滤3次。将得到的催化剂30%fecub保存在无水乙醇中。
分析:由x射线衍射(xrd),透射电子显微镜(tem),与n2物理吸附(bet)对30%fecub样品进行表征。由图1显示,30%fecub样品可观测到清晰的金属铜与氧化亚铜的衍射峰,说明催化剂的活性组分铜处于一种晶态结构。而未发现铁物种的衍射峰,可能由于铁物种的晶粒较小超出了xrd的识别范围,或者是处于一种非晶态结构。通过bet表征,由图2可知,30%fecub样品的比表面积为100.3m2/g。由投射电镜图,图3(a)可观察到催化剂呈细小颗粒状分布,且均匀包裹在另一种浅色物质当中。图3(b)观察到单个催化剂颗粒大约在10nm-40nm之间。
乙酰丙酸加氢活性测试:将0.3g乙酰丙酸,0.1g30%fecub催化剂,15ml1,4-二氧六环,装入高压反应釜聚四氟乙烯内衬中。高压釜密封后以氢气置换5次,以除去釜中的空气。预充氢气后加热至反应温度后恒定氢气压力开始反应,反应温度为200℃,氢气压力5mpa,搅拌速度800转/分,反应时间为6h。反应结束后,冷却至室温,开启反应釜,产物用gc/ms检测。乙酰丙酸转化率100%,1,4戊二醇产率85.1%。余下产物为γ-戊内酯。
实施例2。
催化剂20%fecub的制备:配制包含九水硝酸铁4.04g,三水硝酸铜9.67g与水500ml的盐溶液a。通入氮气保护,放置在冰水浴中,机械剧烈搅拌。配制包含硼氢化钠7.57g,氢氧化钠0.8g与水100ml的还原剂混合液b。将配制好的还原剂混合液b用蠕动泵滴加至盐溶液a中,滴加时间为1.5h.滴加完毕后继续反应2h以等待充分还原。将反应后的产物进行抽滤,用去离子水抽滤5次,无水乙醇抽滤3次。将得到的催化剂20%fecub保存在无水乙醇中。
乙酰丙酸加氢活性测试:将0.3g乙酰丙酸,0.1g20%fecub催化剂,15ml乙醇,装入高压反应釜聚四氟乙烯内衬中。高压釜密封后以氢气置换5次,以除去釜中的空气。预充氢气后加热至反应温度后恒定氢气压力开始反应,反应温度为200℃,氢气压力5mpa,搅拌速度800转/分,反应时间为6h。反应结束后,冷却至室温,开启反应釜,产物用gc/ms检测。乙酰丙酸转化率100%,1,4戊二醇产率42.1%。余下产物为γ-戊内酯与乙酰丙酸乙酯。
实施例3。
催化剂40%fecub的制备:配制包含九水硝酸铁4.03g,三水硝酸铜3.62g与水250ml的盐溶液a。通入氮气保护,放置在冰水浴中,机械剧烈搅拌。配制包含硼氢化钠3.78g,氢氧化钠0.4g与水50ml的还原剂混合液b。将配制好的还原剂混合液b用蠕动泵滴加至盐溶液a中,滴加时间为1.5h.滴加完毕后继续反应2h以等待充分还原。将反应后的产物进行抽滤,用去离子水抽滤5次,无水乙醇抽滤3次。将得到的催化剂40%fecub保存在无水乙醇中。
乙酰丙酸加氢活性测试:将0.3g乙酰丙酸,0.1g40%fecub催化剂,15ml1,4-二氧六环,装入高压反应釜聚四氟乙烯内衬中。高压釜密封后以氢气置换5次,以除去釜中的空气。预充氢气后加热至反应温度后恒定氢气压力开始反应,反应温度为200℃,氢气压力5mpa,搅拌速度800转/分,反应时间为6h。反应结束后,冷却至室温,开启反应釜,产物用gc/ms检测。乙酰丙酸转化率100%,1,4戊二醇产率73.8%。余下产物为γ-戊内酯。
实施例4。
催化剂20%fecub的制备:配制包含九水硝酸铁2.02g,三水硝酸铜4.835g与水250ml的盐溶液a。通入氮气保护,放置在冰水浴中,机械剧烈搅拌。配制包含硼氢化钠3.79g,氢氧化钠0.4g与水50ml的还原剂混合液b。将配制好的还原剂混合液b用蠕动泵滴加至盐溶液a中,滴加时间为1.5h.滴加完毕后继续反应2h以等待充分还原。将反应后的产物进行抽滤,用去离子水抽滤5次,无水乙醇抽滤3次。将得到的催化剂20%fecub保存在无水乙醇中。
乙酰丙酸加氢活性测试:将0.3g乙酰丙酸,0.1g20%fecub催化剂,15ml1,4-二氧六环,装入高压反应釜聚四氟乙烯内衬中。高压釜密封后以氢气置换5次,以除去釜中的空气。预充氢气后加热至反应温度后恒定氢气压力开始反应,反应温度为120℃,氢气压力3mpa,搅拌速度800转/分,反应时间为6h。反应结束后,冷却至室温,开启反应釜,产物用gc/ms检测。乙酰丙酸转化率100%,1,4戊二醇产率0.5%。余下产物为γ-戊内酯(产率>99%)。
本发明还可有其它多种实施例,在不背离本发明精神及其实质的情况下,熟悉本领域的技术人员可根据本发明作出相应的改变和变形,但这些相应的改变和变形都应属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!