衍生检测甾体化合物A环双键异构体的高效液相色谱方法与流程
本发明属于药物分析领域,具体涉及一种衍生测定甾体类化合物a环双键异构体的高效液相色谱方法。
背景技术:
5α-雄甾-2-烯-17-酮在医药领域尤其在甾体激素方面占有重要地位,是合成环硫雄醇、抗心律失常药安律酮和肌肉松弛药泮库溴铵、维库溴铵、哌库溴铵、罗库溴铵等的中间体。
5α-雄甾-2-烯-17-酮作为多种药物的重要中间体,在合成过程中会产生3,4-双键异构体即5α-雄甾-3-烯-17-酮。由于5α-雄甾-3-烯-17-酮与5α-雄甾-2-烯-17-酮的结构与极性非常接近,这个杂质较难通过精制除去或通过后续反应除去。又由于5α-雄甾-3-烯-17-酮与5α-雄甾-2-烯-17-酮的反应位点与活性及其相似,在后续反应中会和相同的反应物产生相似的反应生成更多结构类似的杂质,从而影响下游产品的质量。因此,在后续中间体的质量研究时,需要关注5α-雄甾-2-烯-17-酮与5α-雄甾-3-烯-17-酮的去向与限度。而实验证明,在现有的常规液相色谱条件下,这两种异构体无法实现完全基线分离。
技术实现要素:
在对甾体类化合物进行药物分析的研究过程中,本发明的发明人发现,因为甾体类化合物a环双键异构体相对分子量相等,logp值相等,理化性质类似,尽管采用手性柱和常规的流动相也很难将各成份在同一色谱条件下较好的实现基线分离。如果两个杂质部分交叉无法实现基线分离,则检测结果不准确。对后续研究中异构体的去向与限度无法控制。
为弥补现有技术缺乏分离检测甾体类化合物a环双键异构体技术的不足,本发明的发明人经过大量实验和研究,最终获得一种衍生检测甾体类化合物a环双键异构体的高效液相色谱方法,能够实现甾体类化合物a环双键异构体的有效分离,还能够准确测定异构体的限度,专属性强,灵敏度高;该操作方法简单,具有简便、快速的优点,可用该方法对异构体杂质进行有效的监测和控制。
本发明衍生检测甾体化合物a环双键异构体的高效液相色谱方法,所述甾体化合物a环双键异构体的结构如式i和式ii所示,其中,r1、r2分别独立地选自c1-20的烷基,r3选自h、ac(乙酰基)、tf(三氟甲磺酰基)、bz(苯甲酰基)、ms(甲璜酰基)、ts(对甲苯璜酰基)或c1-20的烷基,该方法包括:将a环双键异构体样品溶解并加入衍生剂,反应结束后挥干溶剂,将反应产物用高效液相色谱进行检测;
上述衍生检测甾体化合物a环双键异构体的高效液相色谱方法,所述衍生剂可以为间氯过氧苯甲酸或n-溴代丁二酰亚胺。本发明一个优选的实施例采用间氯过氧苯甲酸为衍生剂。
进一步地,上述衍生检测甾体化合物a环双键异构体的高效液相色谱方法,所述r1、r2分别独立地选自c1-10的烷基,r3选自h或c1-10的烷基;优选r1、r2分别独立地选自c1-3的烷基,r3为h或c1-3的烷基;更优选r1、r2为甲基,r3为h。
当r1、r2为甲基,r3为h时,所述a环双键异构体为5α-雄甾-2-烯-17-酮和5α-雄甾-3-烯-17-酮,两者结构如下:
上述衍生检测甾体化合物a环双键异构体的高效液相色谱方法,用水、非质子溶剂或者其组合溶解样品,加入衍生剂后室温下反应0.5-3h,然后挥干溶剂,用流动相溶解挥干的样品反应产物,取样10ul用高效液相色谱法进行检测。
甾体化合物a环双键异构体与衍生剂发生反应,生成理化性质差异大的产物,根据产物在液相色谱中的保留时间差,从而实现有效的分离和分析。
本发明所述室温通常是指10~35℃,所述烷基包括支链、直链和环烷基。
所述非质子溶剂包括但不限于乙腈、乙酸乙酯、四氢呋喃、二氯甲烷、二氧六环、丙酮、二甲基甲酰胺和二甲基亚砜中的一种或其组合,更优选为二氯甲烷或二氧六环。
根据本发明一些优选的实施例,单用二氯甲烷或用二氧六环与水的混合物溶解样品。
根据本发明的一些具体实施例,所述样品与衍生剂的摩尔比为1:(1~1.5),优选为1:1。
上述方法中用高效液相色谱检测样品的步骤,所述高效液相色谱的色谱条件为:十八烷基硅烷键合硅胶为填料的色谱柱;采用水-非质子有机溶剂为流动相进行等度洗脱,所述非质子有机溶剂选自乙腈、四氢呋喃或丙酮,优选为乙腈。
根据衍生化后样品溶液的色谱图和样品溶液的色谱图,确定衍生化后样品与样品异构体能达到基线分离。
本发明的检测方法采用单流动相,流动相为水与非质子有机溶剂的混合溶液、十八烷基硅烷键合硅胶为填料的色谱柱,其分离效果符合分析检测的要求,克服了现有技术无法分离甾体类化合物a环双键异构体的难题。
在本发明的一些实施例中,所述流动相为水与乙腈的混合溶液。
所述流动相中水-非质子有机溶剂的体积比为(60-40):(40-60),优选为55:45。
进一步地,高效液相色谱系统进行检测的进样体积10~20μl,等度洗脱的流动相流速为1~2ml/min,优选为1.3ml.min-1,色谱柱的柱温为25~35℃,理论塔板数按5α-雄甾-2-烯-17-酮峰计算不低于2000。
根据本发明的一些具体实施例,高效色谱液相系统的检测器为紫外检测器,检测波长为200nm。
配制的进样溶液,每1ml异构体混合溶液含4mg5α-雄甾-2-烯-17-酮与1mg5α-雄甾-3-烯-17-酮,每1ml供试品溶液含5mg5α-雄甾-2-烯-17-酮。
根据本发明提供的高效液相色谱条件,本领域技术人员可以选择适宜的计算方法计算5α-雄甾-2-烯-17-酮的含量和其异构体5α-雄甾-3-烯-17-酮的限度,所述计算方法包括但不限于内标法、外标法。
本发明的一些实施例采用杂质对照品外标法确定异构体的限度,具体方法如下:
1)配制流动相:以体积比55:45水-乙腈混合物为流动相;
2)衍生:精密称定样品适量置于25ml圆底烧瓶中,向其中精密加入适量二氯甲烷使之溶解,再向其中加入与样品质量比为1:1的间氯过氧苯甲酸。在25℃水浴锅反应1h后将二氯甲烷除尽,再向其中精密加入适量乙腈使之溶解,转移出,即得。
3)单个异构体溶液的制备:
取5α-雄甾-2-烯-17-酮,精密称定,按照步骤2)操作,制成浓度约为5.0mg/ml的样品溶液。
取5α-雄甾-3-烯-17-酮,精密称定,按照步骤2)操作,制成浓度约为5.0mg/ml的样品溶液。
4)分离度溶液的制备:分别取5α-雄甾-2-烯-17-酮与5α-雄甾-3-烯-17-酮,精密称定,按照步骤2)操作,制成每1ml约含5α-雄甾-2-烯-17-酮4.0mg、5α-雄甾-3-烯-17-酮1.0mg的混合溶液。
5)非衍生分离度溶液的制备:分别取5α-雄甾-2-烯-17-酮与5α-雄甾-3-烯-17-酮,精密称定,加乙腈溶解,制成每1ml约含5α-雄甾-2-烯-17-酮4.0mg、5α-雄甾-3-烯-17-酮1.0mg的混合溶液。
6)分别取步骤3)所述单个异构体溶液、步骤4)分离度溶液与步骤5)非衍生分离度溶液10μl注入分离检测系统中,完成异构体的检测。
其中所述高效液相色谱条件如下:
使用以十八烷基硅烷键合硅胶为填料的色谱柱;进行等度洗脱。用高效色谱液相系统进行检测进样体积10μl;流速1~2ml/min;柱温25~35℃;检测波长为200nm。
7)根据步骤6)检测的色谱结果,采用外标法的计算方法计算异构体含量。
根据本发明的发明人研究发现,当衍生剂为间氯过氧苯甲酸时,衍生反应如下所示:
采用其他衍生剂时,适当调整衍生反应溶剂、温度和时间。当衍生剂为n-溴代丁二酰亚胺时,衍生反应如下所示:
试验结果证明本发明衍生检测甾体化合物a环双键异构体的高效液相色谱方法,特别是采用间氯过氧苯甲酸为衍生剂的方法,能较好地分离异构体,方法简便易行,能实现在同一色谱条件下检测双键异构体的含量,更好地监控中间体的质量,同时也为制剂的质量研究提供参考。具体而言,本发明具有以下有益效果:
1、针对现有方法无法有效分离和检测甾体化合物a环双键异构体的不足,本发明填补了a环双键异构体在分析方面的空白;
2、本发明的方法,衍生条件简单,试剂易得,操作简洁,用时较短,具有较好的推广应用前景;
3、高效液相色谱检测条件简单易行,分离效果好、重复性好。
附图说明
图1为本发明实施例1中空白溶液的高效液相色谱图;
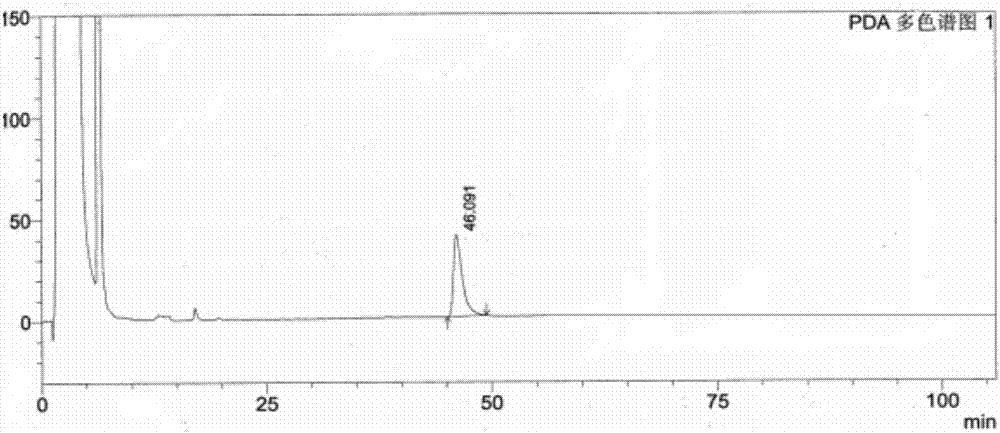
图2为本发明实施例1中5α-雄甾-2-烯-17-酮溶液的高效液相色谱图;
图3为本发明比较例1中5α-雄甾-3-烯-17-酮溶液的高效液相色谱图;
图4为本发明比较例1中分离度溶液的高效液相色谱图;
图5为本发明比较例2中分离度溶液的高效液相色谱图;
图6为本发明比较例3中分离度溶液的高效液相色谱图;
图7为本发明比较例4中分离度溶液的高效液相色谱图;
图8为本发明比较例5中分离度溶液的高效液相色谱图。
具体实施方式
以下将通过优选实施例对本发明的内容进行详细描述。实施例中未注明具体条件的,按照常规条件的实验方法进行。所举实施例是为了更好地对本发明的内容进行说明,但并不能理解为本发明的内容仅限于所举实例。本领域常规技术人员根据上述发明内容对实施方案进行非本质的改进和调整,仍属于本发明的保护范围。
本发明实施例以检测以下结构的甾体类化合物a环双键异构体为例,但不应理解为本发明的方法仅适用于此:
实施例1
仪器与色谱条件:
岛津lc-10ad型高效液相色谱仪;色谱柱为techmatec18-st色谱柱(250mm×4.6mm×5μm);柱温30℃;以水-乙腈(55:45)为流动相;用高效色谱液相系统进行检测,进样体积10μl;流速1.3ml/min;采用紫外检测器;检测波长为200nm。
实验步骤
衍生:精密称定样品适量置于25ml圆底烧瓶中,向其中精密加入适量二氯甲烷使之溶解,再向其中加入与样品摩尔比为1:1的间氯过氧苯甲酸。在25℃水浴锅反应1h后将二氯甲烷除尽,再向其中精密加入适量乙腈使之溶解,转移出,即得。
单个异构体溶液的制备:
取5α-雄甾-2-烯-17-酮,精密称定,按照衍生步骤操作,制成浓度约为5.0mg/ml的样品溶液。
取5α-雄甾-3-烯-17-酮,精密称定,按照衍生步骤操作,制成浓度约为含5.0mg/ml的样品溶液。
分离度溶液的制备:分别取5α-雄甾-2-烯-17-酮与5α-雄甾-3-烯-17-酮,精密称定,按照衍生步骤操作,制成每1ml约含5α-雄甾-2-烯-17-酮4.0mg、5α-雄甾-3-烯-17-酮1.0mg的混合溶液。
分别精密量取空白溶剂、供试品溶液、异构体混合对照品溶液、分离度溶液各10μl,进样测定,记录色谱图,见图1、图2、图3、图4。
试验结果表明,溶剂峰对异构体无干扰,分离度溶液异构体的峰之间的能完全分离。
实施例2
仪器与色谱条件:
岛津lc-10ad型高效液相色谱仪;色谱柱为techmatec18-st色谱柱(250mm×4.6mm×5μm);色谱柱柱温30℃;以水-乙腈(55:45)为流动相;用高效色谱液相系统进行检测进样体积10μl;流速1.5ml/min;采用紫外检测器;检测波长为200nm。
其余操作步骤及条件均同实施例1。
试验结果表明,溶剂峰对异构体无干扰,分离度溶液异构体的峰之间能完全分离。见图5。
实施例3
仪器与色谱条件:
岛津lc-10ad型高效液相色谱仪;色谱柱为techmatec18-st色谱柱(250mm×4.6mm×5μm);柱温35℃;以水-乙腈(55:45)为流动相;用高效色谱液相系统进行检测进样体积10μl;流速1.3ml/min;采用紫外检测器;检测波长为200nm。
其余操作步骤及条件均同实施例1。
试验结果表明,溶剂峰对异构体无干扰,分离度溶液异构体的峰之间能完全分离,见图6。
实施例4
仪器与色谱条件:
岛津lc-10ad型高效液相色谱仪;色谱柱为techmatec18-st色谱柱(250mm×4.6mm×5μm);色谱柱柱温30℃;以水-乙腈(57:43)为流动相;用高效色谱液相系统进行检测进样体积10μl;流速1.3ml/min;采用紫外检测器;检测波长为200nm。
其余操作步骤及条件均同实施例1。
试验结果表明,溶剂峰对异构体无干扰,分离度溶液异构体的峰之间能完全分离,见图7。
实施例5
仪器与色谱条件:
岛津lc-10ad型高效液相色谱仪;色谱柱为techmatec18-st色谱柱(250mm×4.6mm×5μm);色谱柱柱温30℃;以水-乙腈(55::45)为流动相;用高效色谱液相系统进行检测进样体积10μl;流速1.0ml/min;采用紫外检测器;检测波长为200nm。
实验步骤
衍生:精密称定样品适量置于25ml圆底烧瓶中,向其中精密加入适量的二氧六环使之溶解,室温下加入1n的高氯酸水溶液催化,再向其中加入与样品摩尔比为1:1.5的n-溴代丁二酰亚胺。反应过夜,即得
分离度溶液的制备
单个异构体溶液的制备:
取5α-雄甾-2-烯-17-酮,精密称定,按照衍生步骤操作,制成浓度约为5.0mg/ml的样品溶液。
取5α-雄甾-3-烯-17-酮,精密称定,按照衍生步骤操作,制成浓度约为含5.0mg/ml的样品溶液。
分别取5α-雄甾-2-烯-17-酮与5α-雄甾-3-烯-17-酮,精密称定,按照衍生步骤操作,制成每1ml约含5α-雄甾-2-烯-17-酮4.0mg、5α-雄甾-3-烯-17-酮1.0mg的混合溶液。
分别精密量取空白溶剂、供试品溶液、异构体混合对照品溶液、分离度溶液各10μl,进样测定,记录色谱图。
试验结果表明,溶剂峰对异构体无干扰,分离度溶液异构体的峰之间能完全分离,见图8。
根据以上实施例可知,本发明的方法有效解决了因甾体类化合物a环双键异构体用现有方法无法分离而不能进行有效质量监控的问题,当用间氯过氧苯甲酸为衍生剂时,本发明的方法优势更为明显。
尽管已经详细描述了本发明的实施方式,但是应该理解的是,在不偏离本发明的精神和范围的情况下,可以对本发明的实施方式做出各种改变、替换和变更。
- 还没有人留言评论。精彩留言会获得点赞!