一种用液相色谱法分离卡马西平及有关物质的方法与流程
本发明属于医药检测
技术领域:
,具体涉及一种用液相色谱法分离卡马西平及有关物质的方法。
背景技术:
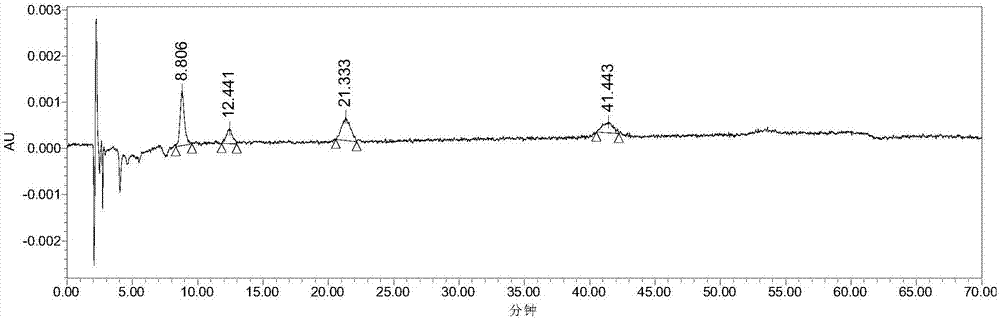
:卡马西平是一种常见精神性药物。临床主要用于:癫痫:部分性发作:复杂部分性发作、简单部分性发作和继发性全身发作。全身性发作:强直、阵挛、强直阵挛发作。三叉神经痛和舌咽神经痛发作,亦用作三叉神经痛缓解后的长期预防性用药。也可用于脊髓痨和多发性硬化,卡马西平别名有:氨甲酰苯卓、氨甲酰苯卓、氨甲酰氮卓、叉颠宁、叉癫宁、得利多、得利益多、芬来普辛、甲酰苯卓、卡巴米嗪、卡巴咪嗪,化学名5H-二苯并[b,f]氮杂卓-5-甲酰胺,CAS号298-46-4。在药物的制备和贮存过程中,需要对合成工艺中可能产生的杂质和降解杂质进行监测。由于这类杂质的化学结构一般与活性成分类似或具渊源关系,故又称为有关物质。因此,实现卡马西平及其有关物质的分离,在卡马西平的合成和制剂过程的质量控制方面具有重要的现实意义。卡马西平药品标准在中国药典2015年版二部第182页有收载,里面记载了卡马西平的有关物质的检查方法,具体为:取本品约50mg,置50mL量瓶中,加甲醇25mL使溶解,用水稀释至刻度,摇匀,作为供试品溶液。精密量取供试品溶液lmL,置50mL量瓶中,用甲醇-水(1:1)稀释至刻度,摇匀,精密量取5mL,置50mL量瓶中,用甲醇-水(1:1)稀释至刻度,摇匀,作为对照溶液。照含量项下的色谱条件和方法,精密量取对照溶液和供试品溶液各20μL,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的6倍。供试品溶液色谱图中如有杂质峰,单个杂质峰面积不得大于对照溶液主峰面积(0.2%),各杂质峰面积的和不得大于对照溶液主峰面积的2.5倍(0.5%)。其具体的液相色谱条件为:以色谱条件与系统适用性试验腈丙基硅烷键合硅胶为填充剂,以甲醇-四氢呋喃-水(120:30:850)为流动相(向1000mL该溶液中加入0.2mL甲酸和0.5mL三乙胺);检测波长230nm。取卡马西平对照品约25mg,置100mL量瓶中,用甲醇-水(1:1)溶解并稀释至刻度,摇匀,取20μL注入液相色谱仪,理论板数按卡马西平计算不低于5000,卡马西平峰与相邻杂质峰的分离度应符合要求。欧洲药典、美国药典和日本药典中也规定了卡马西平的有关物质检查方法,其液相条件与中国药典中相同,不同之处在于,欧洲药典、美国药典和日本药典中列出了卡马西平具体的杂质结构式及部分杂质的含量要求。关于卡马西平的杂质结构式具体情况如下表中所示:由上表可知,日本药典对于卡马西平杂质的规定比较简单,因此,在检测条件相同的情况下,对于卡马西平的杂质种类及杂质量的规定优选美国药典和欧洲药典中的标准。关于卡马西平的中国药典、美国药典、欧洲药典中液相情况及杂质量规定情况如下表中所示:由上述中国药典、美国药典、欧洲药典对于卡马西平对于具体杂质的量的规定可知,卡马西平最重要的杂质为杂质A、杂质B和杂质E,对于其它杂质,不做具体的量的规定和要求。卡马西平的杂质与具体的合成路线有关。卡马西平的常见合成路线(《卡马西平清洁生产工艺研究》)有:路线1:路线2:本公司卡马西平原料药合成路线如下:参考欧洲药典、美国药典和日本药典,查到卡马西平含有杂质A、杂质B、杂质C、杂质D、杂质E、杂质F和杂质G,根据本公司提供合成路线可确定潜在工艺杂质及降解杂质为杂质A、杂质B、杂质C、杂质D和杂质E,其中杂质F和杂质G不是该合成路线产生的。本公司的合成路线中,杂质A、杂质B、杂质C、杂质D和杂质E的具体降解路径如下:杂质A杂质A由合成工艺和降解途径产生:工艺途径:降解途径:杂质B和D杂质B、D:API水解产物及水解产物的异构化产物,可能的反应途径(可通过破坏试验并以LCMS证实):另杂质D也为路线2的起始原料;杂质C杂质C为中间体3与异氰酸反应产物的副产物以及降解杂质;降解途径:卡马西平在酸性条件下降解成杂质A和甲酰胺;在酸性条件下,甲酰胺与卡马西平反应生成杂质C。杂质E杂质E为路线1和厂家提供合成路线起始物料引起的工艺杂质以及降解杂质;降解途径:杂质F杂质F是合成路线1第三部中间体产物。杂质G由本公司提供的合成路线可以看出杂质G不是该合成路线产生的杂质,不对其进行说明解释。根据本公司提供合成路线可确定潜在工艺杂质及降解杂质为杂质A、杂质B、杂质C、杂质D和杂质E,其中杂质F和杂质G不是该合成路线产生的。因此,基于适用于本公司的具体情况的原则,只需要检测杂质A、杂质B、杂质C、杂质D和杂质E即可。发明人在具体试验中发现按照中国药典的液相条件来检测卡马西平杂质,存在下列缺陷:卡马西平每个杂质在9-10μg时的信噪比比较低,在做方法确认时风险比较大,特别是杂质E的灵敏度低。卡马西平杂质E在0.5μg/mL时,积分失败,在2.0μg/mL时,能积出分,但是信噪比太低。ChP、USP和EP供试品溶液浓度为1.0-1.5mg,杂质限度拟定为0.1%左右,限度浓度为1.0-1.5μg/mL。杂质E2μg/mL时的信噪比仅为1.67,说明方法的灵敏度不够。当使用Waterse2695-2489时,杂质峰信噪比改变不大,当杂质浓度为1μg/mL时,杂质E信噪比不能满足验证的条件。因此,需要寻找新的液相条件,来克服上述缺陷。技术实现要素:本发明的目的在于提供一种卡马西平及其有关物质的分离方法。本发明的另一目的在于提供一种卡马西平原料药或其制剂的纯度鉴定方法,以实现卡马西平原料药或其制剂的质量控制。本发明提供了一种用液相色谱法分离卡马西平及有关物质的方法,所述液相色谱法为高效液相色谱法。所述高效液相色谱法包括以下色谱条件:色谱柱填料为腈丙基硅烷键合硅胶;流动相为磷酸二氢铵溶液-乙腈;流动相洗脱方式为线性梯度洗脱;检测器为紫外检测器,进样量为20μL-50μL。所述磷酸二氢铵溶液的浓度为30-60mmol/L。优选的,所述磷酸二氢铵溶液的浓度为50mmol/L。所述流动相线性梯度洗脱条件为:洗脱时间为50-70min,磷酸二氢铵溶液与乙腈的起始体积比为90:10-80:20,磷酸二氢铵溶液与乙腈的终止体积比为80:20-60:40,流动相流速为0.8-1.2ml/min。优选的,所述流动相线性梯度洗脱条件为:磷酸二氢铵溶液与乙腈的起始体积优选为89:11、终止比例为70:30,流动相流速优选为1.0ml/min;洗脱时间优选为65min。所述进样量为50μL。所述紫外检测器的检测波长为215nm。所述卡马西平为卡马西平、卡马西平共晶体、卡马西平中间体;所述有关物质为杂质A~杂质E。本发明的用液相色谱法分离卡马西平及有关物质的方法,所述高效液相色谱法包括以下步骤:(1)色谱条件:用腈丙基硅烷键合硅胶为填充剂,流动相A为50mmol/L磷酸二氢铵溶液,流动相B为乙腈,按下表进行线性梯度洗脱,检测器为紫外检测器,检测波长为215nm;(2)检查方法供试品溶液的制备取卡马西平50mg,精密称定,置50mL量瓶中,加甲醇25mL使溶解,用水稀释至刻度,摇匀,作为供试品溶液;对照溶液的制备精密量取供试品溶液lmL,置50mL量瓶中,用甲醇-水(1:1)稀释至刻度,摇匀,精密量取5mL,置50mL量瓶中,用甲醇-水(1:1)稀释至刻度,摇匀,作为对照溶液;精密量取对照溶液和供试品溶液各50μL,分别注入液相色谱仪,记录色谱图。上述的用液相色谱法分离卡马西平及有关物质的方法,所述方法适用于卡马西平,卡马西平制剂,以及所述卡马西平有关物质杂质A-杂质E各体的分离检测。卡马西平及有关物质的分离效果判定方法及标准为:取卡马西平25mg,精密称定,置100mL量瓶中,用甲醇-水(1:1)稀释至刻度摇匀,取对照溶液20μL注入液相色谱仪,理论塔板数以卡马西平峰计算不低于5000,卡马西平峰与相邻杂质峰的分离度应大于1.7。供试品溶液色谱图中如有杂质峰,杂质A,E:对于每种杂质,不超过对照溶液(a)色谱图中卡马西平的峰面积的一半(0.10%);未指定的杂质:不大于对照溶液(a)色谱图中的卡马西平的峰面积的一半(0.10%);总量:不超过对照溶液(a)色谱图中卡马西平的峰面积的2.5倍(0.5%)。本发明提供的卡马西平及其有关物质的分离方法具有以下优点:1、相对于药典中记载的用液相色谱法分离卡马西平及有关物质的方法,本发明通过改变流动相系统和进样量,能够提高杂质检出的灵敏度,尤其是杂质E的检测灵敏度,从而有效控制卡马西平的质量。2、本发明提供的方法可应用于卡马西平各有关物质个体纯度的检测。由于卡马西平的各有关物质具有与卡马西平相同的化学结构母核,结构非常相似,各有关物质的检测方法与卡马西平的检测方法相同。3、本发明提供的方法,可以完成针对卡马西平及其5个有关物质的有效分离,可以对卡马西平可能存在的有关物质杂质进行全面的检测及控制,也可用于卡马西平及其有关物质的准确定量。4、本发明所述的分离方法,采用通用型色谱柱,方法简单,准确,方便操作。附图说明图1为按照中国药典中规定的色谱条件对卡马西平进行系统适应性实验的液相色谱图。图2为按照中国药典中规定的色谱条件对卡马西平进行线性范围确定实验的液相色谱图,卡马西平杂质浓度为0.5μg/mL。图3为按照中国药典中规定的色谱条件对卡马西平进行线性范围确定实验的液相色谱图,卡马西平杂质浓度为2.0μg/mL。图4为使用不同仪器(Waterse2695-2489)按照中国药典中规定的色谱条件对卡马西平进行了线性范围确定实验色谱图,卡马西平杂质浓度为1.0μg/mL。图5为乙腈-水梯度洗脱方式的色谱条件对卡马西平各杂质的分离度试验,卡马西平杂质浓度为9μg/mL。图6为流动相A0.4%甲酸和0.10%三乙胺溶液;流动相B为乙腈梯度洗脱方式的色谱条件对卡马西平各杂质的分离度试验,卡马西平杂质浓度为9μg/mL。图7为流动梯度相优化后的卡马西平及各杂质的色谱图(流动相A0.4%甲酸和0.10%三乙胺溶液;流动相B为乙腈)。图8为流动梯度相进一步优化后的卡马西平及各杂质的色谱3D图谱(流动相A0.4%甲酸和0.10%三乙胺溶液;流动相B为乙腈)。图9为流动梯度相进一步优化后的卡马西平及各杂质的色谱图(流动相A0.4%甲酸和0.10%三乙胺溶液;流动相B为乙腈)。图10为卡马西平及各杂质的色谱图(流动相A0.4%甲酸和6g/L磷酸二氢钠溶液;流动相B为乙腈)。图11为等度流动相下卡马西平及各杂质的色谱图(流动相A0.4%甲酸和6g/L磷酸二氢钠溶液;流动相B为乙腈)。图12为优化流动相梯度下卡马西平及各杂质的色谱图(流动相A0.4%甲酸和0.10%三乙胺溶液;流动相B为乙腈,检测波长215nm,杂质样品浓度约为9μg/ml)。图13为优化流动相梯度下卡马西平及各杂质的色谱图(流动相A0.4%甲酸和0.10%三乙胺溶液;流动相B为乙腈,检测波长215nm,杂质样品浓度约为0.5μg/ml)。图14为优化流动相梯度下卡马西平及各杂质的色谱图(流动相A50mmol磷酸二氢铵溶液;流动相B为乙腈,检测波长215nm,杂质样品浓度约为5μg/ml)。图15为进一步优化流动相梯度增加进样量条件下卡马西平及各杂质的色谱图(流动相A50mmol磷酸二氢铵溶液;流动相B为乙腈,检测波长215nm,杂质样品浓度约为5μg/ml)。图16为进一步优化流动相梯度增加进样量条件下卡马西平及各杂质的色谱图(流动相A50mmol磷酸二氢铵溶液;流动相B为乙腈,检测波长215nm,杂质样品浓度约为0.5μg/ml)。图17为酸破坏试验中未破坏的卡马西平色谱图(流动相A50mmol磷酸二氢铵溶液;流动相B为乙腈,检测波长215nm)。图18为酸破坏试验中酸破坏的卡马西平色谱图(流动相A50mmol磷酸二氢铵溶液;流动相B为乙腈,检测波长215nm)。图19为碱破坏试验中未破坏的卡马西平色谱图(流动相A50mmol磷酸二氢铵溶液;流动相B为乙腈,检测波长215nm)。图20为碱破坏试验中碱破坏的卡马西平色谱图(流动相A50mmol磷酸二氢铵溶液;流动相B为乙腈,检测波长215nm)。图21为高温破坏试验中未破坏的卡马西平色谱图(流动相A50mmol磷酸二氢铵溶液;流动相B为乙腈,检测波长215nm)。图22为高温破坏试验中高温破坏的卡马西平色谱图(流动相A50mmol磷酸二氢铵溶液;流动相B为乙腈,检测波长215nm)。图23为光照破坏试验中未破坏的卡马西平色谱图(流动相A50mmol磷酸二氢铵溶液;流动相B为乙腈,检测波长215nm)。图24为光照破坏试验中光照破坏的卡马西平色谱图(流动相A50mmol磷酸二氢铵溶液;流动相B为乙腈,检测波长215nm)。图25为本公司卡马西平原料药液相色谱图(批号为2017120501)(流动相A50mmol磷酸二氢铵溶液;流动相B为乙腈,检测波长215nm)。图26为市售样品1卡马西平原料药液相色谱图(流动相A50mmol磷酸二氢铵溶液;流动相B为乙腈,检测波长215nm)。图27为市售样品2卡马西平原料药液相色谱图(流动相A50mmol磷酸二氢铵溶液;流动相B为乙腈,检测波长215nm)。上述图中,所述横坐标的单位为min,纵坐标的单位为AU。具体实施方式下面结合附图和具体实施方式来对本发明作更进一步的说明,以便本领域的技术人员更了解本发明,但并不以此限制本发明。本发明如无特殊说明,浓度一般指的是溶质与溶液的重量体积比g/ml。如果溶质是液体,则浓度是体积比,如0.1%三乙胺水溶液是指0.1ml的三乙胺溶于100ml的水中。所述卡马西平及其有关物质的标准品纯度均为99%以上,由山东则正医药科技有限公司提供;所述化学试剂纯度均为色谱纯级别,分别购自国药集团化学试剂北京有限公司。所述的样品制备方法均为:取卡马西平及杂质A、B、C、D、E杂质,精密称定,用甲醇-水(1:1)稀释至具体实验例和实施例中的浓度。实验例1:药典中色谱条件的验证按照中国药典中规定的色谱条件对卡马西平进行了系统适应性实验,采用的卡马西平样品为本公司生产,批号2017021303。具体色谱条件如下:(1)色谱条件(2)样品制备(3)结果见图1。卡马西平及杂质A、杂质B、杂质C、杂质D和杂质E分离情况如下。名称保留时间面积理论塔板数分离度S/N(信噪比)杂质B9.153439898386158.06杂质A12.37916299435944.7015.93卡马西平14.193850111638022.13725.00杂质C21.20548250835636.0326.41杂质D41.771536433475010.8717.94杂质E54.14814951755354.654.27(4)结论由图1和上述结果可见,卡马西平每个杂质在9-10μg时的信噪比比较低,在做方法确认时风险比较大,特别是杂质E的灵敏度低。按照中国药典中规定的色谱条件对卡马西平进行了线性范围确定实验,采用的卡马西平样品为本公司生产,批号2017021601。具体色谱条件如下:(1)色谱条件(2)当卡马西平杂质浓度为0.5μg/mL时,色谱图见图2。卡马西平及杂质A、杂质B、杂质C、杂质D和杂质E分离情况如下。名称保留时间面积理论塔板数S/N杂质B8.8062750136245.00杂质A12.4411090715351.40杂质C21.3332303639202.14杂质D41.44313035115161.00杂质EN/AN/A(3)当卡马西平杂质为2.0μg/mL时,色谱图见图3。卡马西平及杂质A、杂质B、杂质C、杂质D和杂质E分离情况如下。(4)结论卡马西平杂质E在0.5μg/mL时,积分失败,在2.0μg/mL时,能积出分,但是信噪比太低。原因分析:ChP、USP和EP方法使用的流动相为甲醇:四氢呋喃:水=12:3:85,向1.0L该溶液中加入0.2mL无水甲酸和0.5mL三乙胺,四氢呋喃、三乙胺和甲酸在230nm下有紫外吸收,掩盖了一部分卡马西平各杂质紫外吸收,使得卡马西平各杂质的响应值变低,导致信噪比偏低。ChP、USP和EP供试品溶液浓度为1.0-1.5mg,杂质限度拟定为0.1%左右,限度浓度为1.0-1.5μg/mL。杂质E2μg/mL时的信噪比仅为1.67,说明方法的灵敏度不够。使用不同仪器(Waterse2695-2489)按照中国药典中规定的色谱条件对卡马西平进行了线性范围确定实验。具体色谱条件如下:(1)色谱条件(2)当卡马西平杂质浓度为1μg/mL时,色谱图见图4。(3)结论当使用Waterse2695-2489时,杂质峰信噪比改变不大,当杂质浓度为1μg/mL时,杂质E信噪比不能满足验证的条件。发明人在具体试验中发现按照中国药典的液相条件来检测卡马西平杂质,存在下列缺陷:卡马西平每个杂质在9-10μg时的信噪比比较低,在做方法确认时风险比较大,特别是杂质E的灵敏度低。卡马西平杂质E在0.5μg/mL时,积分失败,在2.0μg/mL时,能积出分,但是信噪比太低。ChP、USP和EP供试品溶液浓度为1.0-1.5mg,杂质限度拟定为0.1%左右,限度浓度为1.0-1.5μg/mL。杂质E2μg/mL时的信噪比仅为1.67,说明方法的灵敏度不够。当使用Waterse2695-2489时,杂质峰信噪比改变不大,当杂质浓度为1μg/mL时,杂质E信噪比不能满足验证的条件。因此,需要寻找新的液相条件,来克服上述缺陷。实施例1:卡马西平有关物质检测条件的探索试验1.首先改变流动相体系,流动相体系中有四氢呋喃,其在低波长处有吸收,可能掩盖了杂质峰,采用乙腈-水梯度洗脱方式。新的色谱条件如下:(1)色谱条件(2)结果色谱图见附图5。卡马西平及各杂质分离情况如下:(3)结论卡马西平各杂质的浓度约为9μg/mL,卡马西平中有一个杂质没有出峰,其它杂质分离良好。原因分析:流动相除去掉四氢呋喃外,同时去掉了三乙胺,而卡马西平杂质含有N,所以不出峰。2.将流动相A改为0.4%甲酸水溶液和0.10%三乙胺水溶液,具体色谱条件如下:(1)色谱条件(2)结果。液相色谱图见附图6。名称保留时间杂质B12.971杂质A17.479杂质C24.903杂质D33.285杂质E35.439(3)结论使用0.4%甲酸和0.10%三乙胺溶液和乙腈作为流动相,在此次梯度下卡马西平各个杂质能完全分离。3.对流动梯度相进行了优化,具体色谱条件如下:(1)色谱条件结果见图7。由附图7可见,杂质C出现在梯度峰上4.进一步优化了流动相梯度,具体色谱条件如下:(1)色谱条件(2)结果见附图8和附图9。(3)结论:杂质C的峰出现在梯度峰上。由3D图谱看出所有杂质最佳吸收波长为215nm。5.将流动相中的盐改为磷酸二氢钠,具体色谱条件如下:(1)色谱条件(2)结果见附图10。(3)结论:流动相中的甲酸和三乙胺改为磷酸氢二钠后,卡马西平杂质峰少一个。6.使用等度流动相(1)色谱条件(2)结果见附图11。(3)结论:杂质E没有出峰。7.优化流动相梯度(1)色谱条件(2)结果见附图12。各杂质及具体分离情况如下。名称保留时间面积理论塔板数S/N杂质B12.6607602985482224.03杂质A18.4407828845526163.65卡马西平20.6132397920055514406.03杂质C29.31611697185167150.88杂质D38.94291508592716358.86杂质E41.932123570060399355.03(3)结论使用三乙胺和甲酸作为流动相中的盐,产生了较大的梯度,而且噪音比较大,信噪比改善不理想。8.优化流动相梯度(1)色谱条件(2)结果见附图13。名称保留时间面积理论塔板数S/N杂质B15.34336948733327.10杂质A18.57740236753524.27杂质C29.58048880842819.86杂质D40.448397874095227.06杂质E46.050530883383928.96(3)结论使流动相得梯度变缓,来改变梯度峰的高度效果不明显。9.流动相中的盐改为磷酸铵盐由于三乙胺末端吸收,在215nm吸收明显,在梯度变化时产生较大的梯度峰,同时对杂质信号也有一定的掩埋。所以选择末端吸收小的磷酸二氢铵进行试验。(1)色谱条件(2)结果见附图14名称保留时间面积理论塔板数分离度S/N杂质B14.2544504864949260.16杂质A18.75248367156575.07234.26卡马西平20.81416804046543381.6652889.68杂质C29.74059852959605.91190.53杂质D40.792584927371559.36320.95杂质E46.697730909266786.04298.09(3)结论流动相A中的盐改为磷酸二氢铵后,卡马西平杂质信噪比得到改善,且梯度峰变的很缓,降低梯度峰上出现未知杂质积分不出来的风险。10.优化流动相梯度,进样量改为50μL,该操作的目的是继续提高信噪比(1)色谱条件(2)结果见附图15和附图16。名称保留时间面积理论塔板数分离度S/N杂质B15.35811308604868627.19杂质A20.674124262350285.28538.16卡马西平23.26826559420127871.5058267.54杂质C34.244177128295885.96624.54杂质D43.8811470892521469.03927.80杂质E48.8811896889337105.57852.55名称保留时间面积理论塔板数分离度S/N杂质B15.495100094449057.02杂质A20.71212372646215.0955.53杂质C34.283156252973810.9862.03杂质D43.878111748505829.2474.81杂质E48.866171804327495.5281.98(3)结论进样量由20μL增加到50μL后,信噪比有了显著的增加,但杂质A和主峰的分离度降低到了1.50,符合条件。用此次的梯度,使得梯度峰变缓,对杂质峰积分影响不大。综合考虑利弊因素,最后使用本次方法的液相条件。最终拟定的卡马西平有关物质色谱条件和方法为:实施例2破坏性试验1酸、碱和氧化破坏(样品组:2017032801)1.1未破坏的原料药取卡马西平原料75mg,精密称定,置50ml量瓶中,加入25ml甲醇溶解,超声使溶解,加水稀释至刻度,摇匀,即得。量取5.00ml溶液于50ml容量瓶中,加入甲醇22ml,加水稀释至刻度,摇匀,即得。结果见附图17:名称保留时间峰名称纯度角度纯度阈值面积面积%分离度s/n121.338主峰0.2320.3455287650310012862.921.2酸破坏酸碱空白:分别移取1mol/L盐酸溶液和氢氧化钠溶液5.00ml于50ml容量瓶中,加入25ml甲醇溶液,加水稀释至刻度,摇匀,即得。取卡马西平原料75mg,精密称定,置50ml量瓶中,加入5ml甲醇溶解,然后加1mol/L盐酸溶液5ml,在80℃水浴条件下加热3h;放冷,加1mol/L氢氧化钠溶液5ml中和后,加入甲醇20ml,超声使溶解,加水稀释至刻度,摇匀,即得。量取5.00ml溶液于50ml容量瓶中,加入甲醇22ml,加水稀释至刻度,摇匀,即得。结果见附图18:名称保留时间峰名称纯度角度纯度阈值面积面积%分离度s/n115.105杂质B1.1551.292531900.1123.69218.721杂质A1.7381.768325050.063.5815.93321.237主峰0.1360.3204595250691.682.3316864.60442.030杂质D0.1860.24740429058.0720.992155.77552.271未知杂质8.0807.669443320.098.2113.4结论:卡马西平在甲醇的酸性溶液中,产生降解,各降解杂质与主峰以及已知杂质的分离度大于1.5;主峰的纯度角小于纯度阈值,合格。1.3碱破坏取卡马西平原料75mg,精密称定,置50ml量瓶中,加入10ml甲醇溶解,然后加1mol/L氢氧化钠溶液5ml,在80℃水浴条件下加热3h;放冷,加1mol/L盐酸溶液5ml中和后,加入甲醇15ml,超声使溶解,加水稀释至刻度,摇匀,即得。量取5.00ml溶液于50ml容量瓶中,加入甲醇22ml,加水稀释至刻度,摇匀,即得。结果见附图19:名称保留时间峰名称纯度角度纯度阈值面积面积%分离度s/n121.126主峰0.1730.3515125655998.5812913.80241.940杂质D0.4550.3697381511.4220.6271.79结论:卡马西平在甲醇的碱性溶液中,产生降解,降解杂质与主峰的分离度大于1.5;主峰纯度角小于纯度阈值。1.4氧化破坏氧化空白:移取30%双氧水溶液5.00ml于50ml容量瓶中,加入25ml甲醇溶液,加水稀释至刻度,摇匀,即得。取卡马西平原料75mg,精密称定,置50ml容量瓶中,加入5ml甲醇溶解,然后加30%双氧水溶液5ml,在80℃水浴条件下加热3h;放冷,加入甲醇20ml,超声使溶解,加水稀释至刻度,摇匀,即得。量取5.00ml溶液于50ml容量瓶中,加入甲醇22ml,加水稀释至刻度,摇匀,即得。结果附图20。名称保留时间峰名称纯度角度纯度阈值面积面积%分离度s/n15.645未知杂质13.6614.43674020.017.6228.222未知杂质4.8895.097288480.066.1119.4139.299未知杂质3.9863.903579490.111.7326.09410.23未知杂质47.0715.776421930.081.286.53513.384未知杂质18.77914.806199290.044.088.18618.724杂质A2.462.288203590.045.746.91721.048主峰0.1550.3535075065799.652.1113661.60结论:卡马西平在甲醇的双氧水溶液中,产生降解,各降解杂质与主峰以及已知杂质的分离度大于1.5;主峰的纯度角小于纯度阈值,合格。1.5物料守恒计算公式:A1=原料药峰面积;A2=样品峰面积和;m1=原料药质量;m2=样品质量;2高温破坏(样品组:2017031702)2.1未破坏原料药取卡马西平原料75mg,精密称定,置50ml量瓶中,加入25ml甲醇溶解,超声使溶解,加水稀释至刻度,摇匀,即得。量取5.00ml溶液于50ml容量瓶中,加入甲醇22ml,加水稀释至刻度,摇匀,即得。结果见附图21:名称保留时间纯度角度纯度阈值面积面积%分离度122.2150.210.309521038431002.2高温破坏取经130℃烘箱中加热4h后的卡马西平原料75mg,精密称定,置50ml容量瓶中,加25ml甲醇,加水稀释至刻度,摇匀,即得。量取5.00ml溶液于50ml容量瓶中,加入甲醇22ml,加水稀释至刻度,摇匀,即得。结果见附图22:名称保留时间纯度角度纯度阈值面积面积%分离度s/n122.1750.2010.3025266422910012932.07结论:在130℃下,能比较稳定的存在;主峰的纯度角小于纯度阈值。2.3物料守恒计算公式:A1=原料药峰面积;A2=样品峰面积和;m1=原料药质量;m2=样品质量;3光照破坏(样品组:2017032701)3.1未破坏原料药取卡马西平原料75mg,精密称定,置50ml量瓶中,加入25ml甲醇溶解,超声使溶解,加水稀释至刻度,摇匀,即得。量取5.00ml溶液于50ml容量瓶中,加入甲醇22ml,加水稀释至刻度,摇匀,即得。结果见附图23:名称保留时间纯度角度纯度阈值面积面积%分离度121.6820.1450.371528782431003.2光照破坏取经照度4500lx,近紫外能量≥200w·hr/m2下照射13天卡马西平原料75mg,精密称定,置50ml容量瓶中,加25甲醇,超声使溶解,加水稀释至刻度,摇匀,即得。量取5.00ml溶液于50ml容量瓶中,加入甲醇22ml,加水稀释至刻度,摇匀,即得。结果见附图24:名称保留时间纯度角度纯度阈值面积面积%分离度s/n121.3930.1520.3625316213410016061.61结论:在白光和紫外照射下,卡马西平能稳定存在;主峰的纯度角小于纯度阈值。3.3物料守恒计算公式:A1=原料药峰面积;A2=样品峰面积和;m1=原料药质量;m2=样品质量;实施例3卡马西平原料药的有关物质检查待测样品为山东则正医药技术有限公司生产的卡马西平原料药(批号为2017120501)、市售样品两批,分别为市售样品1、市售样品2。检测步骤如下:供试品溶液的制备取卡马西平50mg,精密称定,置50mL量瓶中,加甲醇25mL使溶解,用水稀释至刻度,摇匀,作为供试品溶液;对照溶液的制备精密量取供试品溶液lmL,置50mL量瓶中,用甲醇-水(1:1)稀释至刻度,摇匀,精密量取5mL,置50mL量瓶中,用甲醇-水(1:1)稀释至刻度,摇匀,作为对照溶液;精密量取对照溶液和供试品溶液各50μL,分别注入液相色谱仪,检测结果如图25,图26,图27,经计算结果见下表。有关物含量批号2017120501市售样品1市售样品2标准规定杂质A0.020.050.05<0.10%杂质B未检出未检出未检出<0.10%杂质C0.070.11未检出<0.10%杂质D未检出未检出未检出<0.10%杂质E未检出未检出未检出<0.10%总杂质0.090.820.81<0.50%结果显示:本公司生产的马西平样品的纯度为99.91%,检测结果符合质量标准要求。市售样品纯度分别为99.18%、99.19%,检测结果不符合质量标准要求。本方法可有效应用于卡马西平原料药的质量控制。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1