一种规模化的核心岩藻糖基化修饰位点占有率定量方法与流程
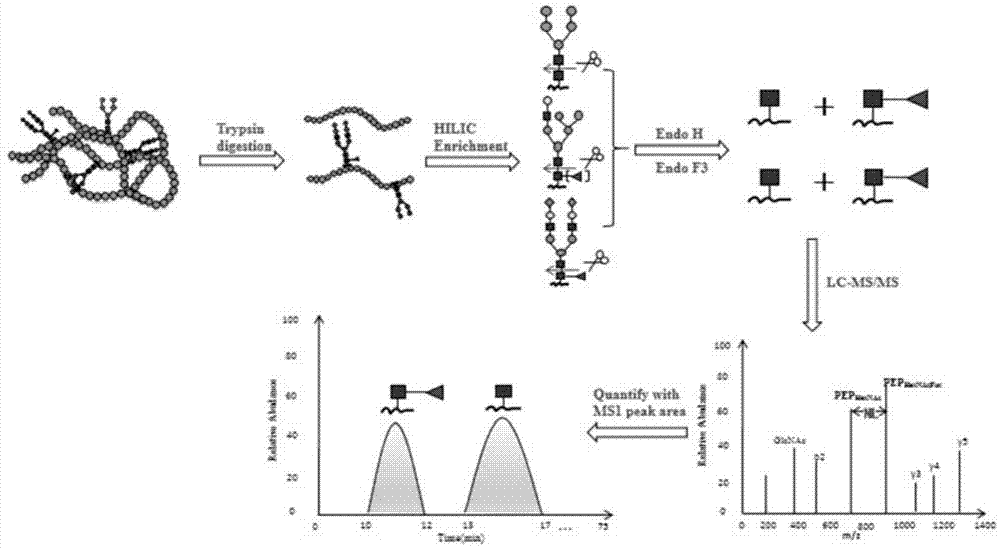
本发明涉及一种规模化的核心岩藻糖基化修饰位点占有率定量方法。
背景技术
蛋白质核心岩藻糖基化修饰(corefucosylation,cf)是一种特殊的n-连接糖基化修饰。核心岩藻糖基化的改变是恶性肿瘤发生发展过程中重要的特征之一。对于蛋白质核心岩藻糖基化修饰鉴定的文章屡见不鲜,但是这些报道对于核心岩藻糖基化修饰的研究都是停留在定性鉴定方面,对于核心岩藻糖修饰位点特异性占有率的定量研究,仍然存在很大的挑战和发展空间。曾有报道一种鉴定和定量核心岩藻糖基化修饰的方法,该方法是将不同的凝集素(lca,psa,aal,ltl,ueai和aol)与商业化的max柱进行单一的或串联的结合,再将富集纯化得到的含有cf修饰的肽段使用同位素辅助多重定量试剂(tmt)进行标记,运用液质联用进行检测,并用gpqeust软件进行数据分析处理,在前列腺癌样本中鉴定到252个核心岩藻糖基化修饰蛋白并定量了973个核心岩藻糖基化修饰的糖肽。但该方法存在以下不足:1)采用凝集素富集含核心岩藻糖修饰的糖肽,丢失了非核心岩藻糖修饰糖肽,从而无法实现位点特异的核心岩藻糖基化修饰占有率定量分析;2)tmt等标记试剂价格昂贵,一次最多只能标记10例样本,无法满足大规模样本的分析;3)实验操作时间较长,重复性不佳。4)从完整糖肽的水平进行质谱分析困难极大,这是由于糖链结构复杂,且糖肽质量数较高,一般质谱方法无法得到完整详细的糖肽谱图;5)糖基化修饰的微不均一性和结构异质性使得谱图的解析和数据处理带来挑战。
技术实现要素:
本发明的目的是提供一种规模化的核心岩藻糖基化修饰位点占有率定量方法。
本发明要求保护一种内切糖苷酶在蛋白质核心岩藻糖化修饰鉴定和/或测定位点占有率中的应用。
上述应用中,所述内切糖苷酶为endoh和/或endof3;
所述内切糖苷酶endoh的酶切范围为ph5.0-6.0;所述内切糖苷酶endof3的酶切范围为ph4.0-5.0;
适用的蛋白质糖基化修饰类型为n-连接糖基化修饰。
本发明还要求保护一种蛋白质核心岩藻糖化修饰鉴定与位点占有率大规模定量的方法,该方法包括如下步骤:
1)从待测样品中分离得到糖肽混合物,再用内切糖苷酶进行酶切,将糖肽糖链部分的外围天线全部切除,剩余产物为连接有一个最内侧glcnac的肽段和连接有一个最内侧的glcnac+fuc的肽段的混合物,记为简化糖肽样本;所述glcnac记为hn;所述glcnac+fuc记为cf;
所述连接有glcnac的肽段记为hn肽段;
所述连接有glcnac+fuc的肽段记为cf的肽段;
2)采用色谱-质谱联用方法对步骤1)所得简化糖肽样本进行检测,得到质谱源文件;
3)将步骤2)所得质谱源文件进行谱图筛选,得到所述cf的肽段和所述hn肽段的谱图,再对cf上述谱图进行定性分析,得到所述简化糖肽的定性结果及scannumber;
所述谱图筛选的规则包括:
先提取所述步骤2)所得质谱源文件中含有糖特征碎片的信号离子的谱图,再筛查所得谱图中是否核心岩藻糖特征的中性丢失母离子;
若筛查到谱图中含有核心岩藻糖特征的中性丢失母离子,则对谱图进行母离子重校正并删除糖特征碎片离子峰;
所述母离子重校正所用公式如下:重校正后的母离子质量=原始母离子质量-核心岩藻糖质量;
所述母离子重校正包括:将原始母离子峰的质量数减去一个核心岩藻糖的质量数;
所述删除糖特征碎片离子峰步骤中,删除的糖特征碎片离子峰对应的质量数为126、138、168、186和204;
所述符合n-糖基化特征序列为(nx(s/t/c),x不为p);x代表不为p的氨基酸;
所述scannumber为谱图鉴定序列号;
4)根据步骤3)所得scannumber,在步骤2)所得质谱源文件进行匹配,得到同一位点上hn肽段或cf的肽段的rt值和理论质量数;并根据质量差和保留时间差的规则,利用已获得的hn肽段或cf的肽段的rt值和理论质量数去推测同一位点上cf的肽段或hn肽段的rt值和理论质量数;
所述质量差规则为hn肽段和cf的肽段的质量数相差一个核心岩藻糖的质量数;
所述保留时间差的规则为hn肽段和cf的肽段的保留时间差在1min内;
5)根据步骤4)所得两类简化糖肽的rt值和理论质量数,在ms1文件中提取对应的ms1峰面积,再根据公式:
cf的ms1峰面积除以hn的ms1峰面积与cf的ms1峰面积之和,得到特定糖基化位点上核心岩藻糖基化修饰的位点占有率;
所述名为ms1文件按照包括如下步骤的方法生成:将步骤2)所得质谱源文件中一级质谱图的所有信息进行提取而得;所述所有信息包括:所有一级母离子的精确质量数、保留时间和峰面积。
上述方法的步骤1)中,所述糖肽混合物按照如下步骤制得:
对蛋白质混合物进行酶切,得到肽段混合物,再进行糖肽混合物的富集;
具体的,所述蛋白质混合物可为从离体的细胞、组织、尿液或血清等生物样品中提取而得;
从细胞、组织中提取蛋白混合物的方法具体可为:利用细胞裂解液将细胞或组织混悬后,冰上超声提取蛋白混合物;从尿液中提取蛋白混合物的方法为丙酮沉淀法;从酵母细胞中提取蛋白质混合物的方法为玻璃珠法;
所述酶切步骤中,酶切方法为fasp酶切、溶液酶切或胶内酶切方式;
酶切所用酶选自trypsin、gluc、chymotrypsin和lys-c中至少一种;
所述trypsin的反应条件包括:50mmnh4hco3、ph7.8;
所述gluc的反应条件包括:ph4.0-9.0;
所述chymotrypsin的反应条件包括:ph7.0-9.0;
所述lys-c的反应条件包括ph7.0-9.0。
所述酶切步骤包括:将提取的蛋白混合物缓冲液用8mua(ph8.0-8.5)替换变性,分批次加入50mmdtt(37度)和20mmiaa(室温)进行还原烷基化,再用ph8.0、50mmnh4hco3替换缓冲液,最后按照比例(酶:蛋白=1:50)加入相应的酶,37度恒温过夜
所述内切糖苷酶为endoh和/或endof3;
所述内切糖苷酶endoh的酶切范围为ph5.0-6.0;所述内切糖苷酶endof3的酶切范围为ph4.0-5.0。
所述富集步骤中,富集方法为常规方法,具体可为采用亲水相互作用色谱(hydrophilicinterationliquidchromatography,hilic)富集;具体为一步hilic;具体条件如下:称取5mghilic填料于离心管,加入适量体积0.1%tfa,放置于混悬仪上慢速摇动15min后取下离心管,在掌上离心机上短暂离心,用移液枪吸除上层液体(避免吸入下层hilic填料),再加入bbh(80:20:0.2=乙腈:双蒸水:tfa),放置于混悬仪上慢摇15min,并重复此步骤3次;将用bbh复溶的酶切样品用移液枪转移至含有填料的离心管中,放置于混悬仪上慢速摇动2h,让填料与样品充分结合;将悬浮有hilic填料的混合液利用移液枪加入到填有c8筛板的枪头中,利用注射器将上层溶液打下,从而使糖肽保留在hilic小柱上;加入bbh的溶液清洗hilic上非特异性吸附的非极性肽段,重复2次;再用0.1%tfa洗脱糖肽2次,合并热干。
所述步骤2)中,在所述检测步骤之前,对所述简化糖肽样本进行如下预处理:热干脱盐,再用甲酸的水溶液复溶;所述甲酸的水溶液中,甲酸与水的体积比具体为0.1:99.9;
所述热干脱盐的具体步骤包括:
a、将tip小柱填一层c18膜,然后分别用a液、b液和c液活化,得到活化后的tip小柱;
b、用b液复溶所述简化糖肽样本,加入到所述活化后的tip小柱中,加入c液过膜脱盐,再b液进行洗脱,热干;
所述a液为乙腈;
所述b液为由体积比为50:50:0.05的乙腈、双蒸水和tfa组成的混合液;
所述c液为体积百分浓度为0.1%的tfa溶液;
所述检测步骤中,色谱部分的检测条件如下:
流动相a相为甲酸的水溶液;所述甲酸的水溶液中,甲酸与水的体积比具体为0.1:99.9;
b相为由体积比为99.9:0.1的乙腈(acn)和甲酸(fa)组成的混合液;
毛细管色谱柱的内径为150μm;柱长120mm;色谱柱填料为粒径为1.9μm的c18;
梯度洗脱的条件如下:
0min:从5%b相开始;
1min-13min末:从5%b相升至10%b相;
14min起-50min末:从10%b相升至22%b相;
51min起-67min末:从22%b相升至36%b相;
68min起-68min末:从36%b相升至100%b相;
69min起-75min末:维持100%b相;
流速为600nl/min;
质谱部分的检测条件如下:采用正离子数据依赖性(dda)扫描模式,nce(归一化碰撞能量)值设为27,一级全扫描范围(ms1)设为m/z300-2000,自动增益设置为3e6,分辨率为120000,最大离子注入时间为80ms。一级全扫描中top20的离子进行二级碎裂扫描,二级自动增益设为5e4,最大离子注入时间为45ms。
所述步骤3)中,所述谱图筛选具体可先将步骤2)所得质谱源文件转换成mgf格式,运用perl语言编写的程序包按照所述谱图筛选的规则对其进行谱图筛选;定性分析的方法为数据处理;
所述数据处理具体包括:用字母“j”替换所有背景数据库中符合n-糖基化特征序列的n,再利用pfind搜库。
用字母“j”替换所有背景数据库中符合n-糖基化特征序列(nx(s/t/c),x不为p)的“n”的目的是将“j”定义为与天冬酰胺(n)具有相同分子量的氨基酸;
更具体的,用pfind2.8.0.1生产对应的反库,采用正反库(target-decoy)方法进行数据库检索。该数据库检索时,设置可变修饰:蛋氨酸氧化(oxidation_met)、蛋白质n末端乙酰化(acetylation_protein_n_terminal)以及j残基的203.079da的质量改变(_glcnac_j_203.079),固定修饰设置为:半胱氨酸烷基化(carbamidomethylation_cys)。最多允许2个trypsin漏切位点,母离子误差10ppm,子离子误差15mmu,可容许1%的肽段水平假阳性率(fdr)
所述步骤4)中,所述将步骤2)所得质谱源文件中一级质谱图的所有信息进行提取具体可按照如下步骤进行:利用proteinwizard程序中msconvert模块将步骤2)所得raw格式的质谱源文件文件转换成mzxml格式,再利用r语言程序包中的readmzxmlpackage,从mzxml文件中进行信息提取。
本发明提供的快速、规模化的蛋白质核心岩藻糖化修饰鉴定与位点占有率大规模精确定量方法,是先将蛋白通过胰蛋白酶水解得到糖肽和非糖肽,然后利用亲水相互作用色谱(hilic)富集糖肽的特性除去多余的非糖肽部分及其它一些杂质,再通过糖苷内切酶h(endoh)和糖苷内切酶f3(endof3)分别进行酶切或者联合酶切,将糖肽糖链部分的外围天线全部切除,只留下肽段连接一个最内侧的n-乙酰葡糖胺(glcnac)和肽段连接一个最内侧的n-乙酰葡糖胺加一个核心岩藻糖(glcnacfuc)的简化糖肽的结构。这种简化的糖肽形式同时保留了核心岩藻糖的结构及肽段序列信息,又降低了糖肽的复杂程度,更有利于被质谱所检测,也将为后续的数据定量分析带来极大的便利。本方法中同时可得到只含有一个n-乙酰葡糖胺以及含有一个n-乙酰葡糖胺加核心岩藻糖的修饰,即hn结果以及cf结果。同时按照以上数据处理方法,即根据保留时间相近、质量数差异一个岩藻糖残基的特征,利用hn修饰肽段的鉴定结果,推测相近rt时间内cf的结果,反之亦然,基于上述成对数据,可计算获得位点特异性cf修饰的比值,得到定量结果。该方法简化了糖肽质谱分析,并能够对核心岩藻糖基化修饰的位点占有率进行定量分析。
附图说明
图1为方法的技术路线图;
图2为数据处理流程图;
图3为标准品ms1峰面积质谱提取图;
图4为yeast假阳性结果质谱ms2图谱;
图5为肝癌细胞定量数据相关性分析图。
图6为肝癌细胞定量数据cf-ratio特征分析图。
具体实施方式
下面结合具体实施例对本发明作进一步阐述,但本发明并不限于以下实施例。所述方法如无特别说明均为常规方法。所述原材料如无特别说明均能从公开商业途径获得。
下述实施例中,用hilic富集填料的tip小柱进行富集得到糖肽混合物的具体条件如下:称取5mghilic填料于离心管,加入适量体积0.1%tfa,放置于混悬仪上慢速摇动15min后取下离心管,在掌上离心机上短暂离心,用移液枪吸除上层液体(避免吸入下层hilic填料),再加入bbh(80:20:0.2=乙腈:双蒸水:tfa),放置于混悬仪上慢摇15min,并重复此步骤3次;将用bbh复溶的酶切样品用移液枪转移至含有填料的离心管中,放置于混悬仪上慢速摇动2h,让填料与样品充分结合;将悬浮有hilic填料的混合液利用移液枪加入到填有c8筛板的枪头中,利用注射器将上层溶液打下,从而使糖肽保留在hilic小柱上;加入bbh的溶液清洗hilic上非特异性吸附的非极性肽段,重复2次;再用0.1%tfa洗脱糖肽2次,合并热干。
下述实施例中,热干脱盐的具体步骤包括:
a、将tip小柱填一层c18膜,然后分别用a液、b液和c液活化,得到活化后的tip小柱;
b、用b液复溶所述简化糖肽样本,加入到所述活化后的tip小柱中,加入c液过膜脱盐,再b液进行洗脱,热干;
所述a液为乙腈;
所述b液为由体积比为50:50:0.05的乙腈、双蒸水和tfa组成的混合液;
所述c液为体积百分浓度为0.1%的tfa溶液。
实施例1、标准品rnaseb和igg(免疫球蛋白g)
运用fasp酶切方式将称取的标准品(标准蛋白rnaseb)(8m尿素溶解)用trypsin酶进行酶切得到肽段混合物,然后通过运用hilic富集填料的tip小柱进行富集得到糖肽混合物;再先后使用糖苷内切酶endoh(ph5.0-6.0)和endof3(ph4.0-5.0)进行酶切,将糖肽糖链部分的外围天线全部切除,剩余产物为连接有一个最内侧n-乙酰葡糖胺glcnac的肽段和连接有一个最内侧的n-乙酰葡糖胺加一个核心岩藻糖glcnac+fuc的肽段的混合物,记为简化糖肽样本;所述glcnac记为hn;所述glcnac+fuc记为cf;连接有glcnac的肽段记为hn肽段;连接有glcnac+fuc的肽段记为cf的肽段;将样品热干脱盐后送入液质联用检测。
液质联用部分方法为:
a.液相部分:流动相a相为0.1%fa溶液,b相为acn-fa(99.9:0.1,v/v)溶液,毛细管色谱柱150μm×120mmc18(填料粒径1.9μm),梯度洗脱的条件如下:
0min:从5%b相开始;
1min-13min末:从5%b相升至10%b相;
14min起-50min末:从10%b相升至22%b相;
51min起-67min末:从22%b相升至36%b相;
68min起-68min末:从36%b相升至100%b相;
69min起-75min末:维持100%b相;
流速为600nl/min。
b.质谱部分:采用正离子数据依赖性(dda)扫描模式,nce(归一化碰撞能量)值设为27,一级全扫描范围(ms1)设为m/z300-2000,自动增益设置为3e6,分辨率为120000,最大离子注入时间为80ms。一级全扫描中top20的离子进行二级碎裂扫描,二级自动增益设为5e4,最大离子注入时间为45ms。得到质谱raw文件。
定性数据处理:运用proteomediscoverer2.1软件(美国,thermofisher公司)将raw文件转换为mgf格式,默认参数设置。运用perl语言编写的程序包(对应软件为perl软件)对转换后的mgf格式文件进行谱图挑选,挑选原则为对含有糖特征碎片的信号离子进行谱图提取,进而对谱图中核心岩藻糖中性丢失母离子进行筛查,若检测到中性丢失母离子,则对候选谱图进行母离子重校正以及糖特征碎片离子去除。然后将处理得到的文件利用pfind2.8.0.1软件进行数据库检索。使用homosapiensuniprotdatabase(人)(20205个条目)作为背景数据库。用字母“j”替换所有背景数据库中符合n-糖基化特征序列(nx(s/t/c),x不为p)的“n”,目的是将“j”定义为与天冬酰胺(n)具有相同分子量的氨基酸。用pfind2.8.0.1生产对应的反库,采用正反库(target-decoy)方法进行数据库检索。该数据库检索时,设置可变修饰:蛋氨酸氧化(oxidation_met)、蛋白质n末端乙酰化(acetylation_protein_n_terminal)以及j残基的203.079da的质量改变(_glcnac_j_203.079),固定修饰设置为:半胱氨酸烷基化(carbamidomethylation_cys)。最多允许2个trypsin漏切位点,母离子误差10ppm,子离子误差15mmu,可容许1%的肽段水平假阳性率(fdr)。
定量数据处理:首先将raw文件利用proteinwizard软件中msconvert模块转换为mzxml格式,再利用r语言中的readmzxmlpackage,从mzxml文件中提取ms1完整信息(包括实际保留时间值(rt)、对应的谱图序号以及一级峰强度)ms1.txt,另一方面利用搜库结果进行数据处理得到简化糖肽的scannumber和m/z信息。利用此信息,结合ms1.txt文件中的详细信息,得到该糖肽的rt推测值以及理论质量数,并根据质量差和保留时间差的规则,利用已获得的hn肽段或cf的肽段的rt值和理论质量数去推测同一位点上cf的肽段或hn肽段的rt值和理论质量数;从而进一步通过rt推测值提取ms1的峰面积作为该糖肽的定量值信息。
质量差规则为hn肽段和cf的肽段的质量数相差一个核心岩藻糖的质量数;
保留时间差的规则为hn肽段和cf的肽段的保留时间差在1min内;
标准蛋白rnaseb仅有高甘露型的n-糖链,不含核心岩藻糖修饰;igg既含有高甘露糖型,也含有复合型和杂合型的n-糖链。标准蛋白rnaseb上主要糖基化肽段为nltk,分别提取了nltk肽段连接一个n-乙酰葡糖胺和nltk肽段连接一个n-乙酰葡糖胺加一个核心岩藻糖的简化糖肽的离子的一级质谱图(图3中左图);igg主要糖基化肽段为eeqynstyr,同样分别提取了eeqynstyr肽段连接一个n-乙酰葡糖胺和eeqynstyr肽段连接一个n-乙酰葡糖胺加一个核心岩藻糖的简化糖肽的一级质谱图(图3中右图),可以看到经endoh和endof3连续酶切后,标准蛋白rnaseb可以得到肽段连接一个n-乙酰葡糖胺(pep-glcnac)的简化糖肽的结构,而由于其不含核心岩藻糖修饰,所以提取不到肽段连接一个n-乙酰葡糖胺加一个核心岩藻糖(pep-glcnacfuc)的简化糖肽的一级质谱图;而igg则同时可以得到肽段连接一个n-乙酰葡糖胺(pep-glcnac)和肽段连接一个n-乙酰葡糖胺加一个核心岩藻糖(pep-glcnacfuc)的简化糖肽的一级质谱图。
以上结果证实了糖苷内切酶h(endoh)和糖苷内切酶f3(endof3)酶切的特异性,并为进一步用于复杂样本实验提供了理论基础。并且通过ms结果可以看到,简化糖肽pep-glcnacfuc结构(即含cf糖肽肽段)的亲水性高于pep-glcnac结构(即仅含hn肽段),从而先于pep-glcnac结构出峰,并且两种简化糖肽形式保留时间差在1min内,这也是为后续定量数据处理中hn结果与cf结果保留时间互相推测的条件提供事实依据。
实施例2:新鲜酿酒酵母细胞
玻璃珠法提取新鲜酿酒酵母细胞(培养基为ypd)得到蛋白混合物,运用fasp酶切方式提取的蛋白混合物用trypsin酶进行酶切得到肽段混合物,然后通过运用hilic富集填料的tip小柱进行富集得到糖肽混合物;再先后使用糖苷内切酶endoh(ph5.0-6.0)和endof3(ph4.0-5.0)进行酶切,剩余产物为连接有一个最内侧n-乙酰葡糖胺glcnac的肽段和连接有一个最内侧的n-乙酰葡糖胺加一个核心岩藻糖glcnac+fuc的肽段的混合物,记为简化糖肽样本;所述glcnac记为hn;所述glcnac+fuc记为cf;连接有glcnac的肽段记为hn肽段;连接有glcnac+fuc的肽段记为cf的肽段;将样品热干脱盐后送入液质联用检测。
液质联用部分方法为:
a.液相部分:同实施例1;
b.质谱部分:同实施例1;
定性数据处理:运用proteomediscoverer2.1软件(美国,thermofisher公司)将raw文件转换为mgf格式,默认参数设置。运用perl语言编写的程序包对转换后的mgf格式文件进行谱图挑选,挑选原则为对含有糖特征碎片的信号离子进行谱图提取,进而对谱图中核心岩藻糖中性丢失母离子进行筛查,若检测中性丢失母离子,则对候选谱图进行母离子重校正以及糖特征碎片离子去除。然后将处理得到的文件利用pfind2.8.0.1软件进行数据库检索。使用uniprotdatabaserelease_20170916(酵母)(6721个条目)作为背景数据库。用字母“j”替换所有背景数据库中符合n-糖基化特征序列(nx(s/t/c),x不为p)的“n”,目的是将“j”定义为与天冬酰胺(n)具有相同分子量的氨基酸。用pfind2.8.0.1生产对应的反库,采用正反库(target-decoy)方法进行数据库检索。该数据库检索时,设置可变修饰:蛋氨酸氧化(oxidation_met)、蛋白质n末端乙酰化(acetylation_protein_n_terminal)以及j残基的203.079da的质量改变(_glcnac_j_203.079),固定修饰设置为:半胱氨酸烷基化(carbamidomethylation_cys)。最多允许2个trypsin漏切位点,母离子误差10ppm,子离子误差15mmu,可容许1%的肽段水平假阳性率(fdr)。
定量数据处理:首先将raw文件利用proteinwizard软件中msconvert模块转换为mzxml格式,再利用r语言中的readmzxmlpackage,从mzxml文件中提取ms1完整信息(包括实际保留时间值(rt)、对应的谱图序号以及一级峰强度)ms1.txt,另一方面对搜库结果进行数据处理得到简化糖肽的scannumber和m/z信息。利用此信息,结合ms1.txt文件中的详细信息,得到该糖肽的rt推测值以及理论质量数,并根据质量差和保留时间差的规则,利用已获得的hn肽段或cf的肽段的rt值和理论质量数去推测同一位点上cf的肽段或hn肽段的rt值和理论质量数;从而进一步通过rt推测值提取ms1的峰面积作为该糖肽的定量值信息。
质量差规则为hn肽段和cf的肽段的质量数相差一个核心岩藻糖的质量数;
保留时间差的规则为hn肽段和cf的肽段的保留时间差在1min内;
为了验证所建立的核心岩藻糖修饰鉴定与定量方法的特异性,又以酵母细胞全蛋白质为实验对象,做了三次生物学平行重复实验,实验结果通过搜库,鉴定到的结果如表1所示。因为酵母细胞全蛋白不含核心岩藻糖修饰,而三次平行实验鉴定的结果中有两例鉴定到含1个带cf修饰的肽段,运用搜库时限定了1%的fdr值,这也与实验预期相符合。由于简化糖肽的形式下,核心岩藻糖与n-乙酰葡糖胺之间的糖苷键会最先断裂,母离子会产生146da(核心岩藻糖残基分子量)的质量亏损,其特征谱图会形成母离子和质量差为146da的中性丢失所构成的离子对,即为典型的含核心岩藻糖修饰的简化糖肽的2电荷的质谱图(图4中c)。为了验证结果是否可靠,将检出的含cf修饰的肽段ndtlssflnr(图4中a)和sespfsafatnitkpssttpafsfgnstmjk(图4中b)分别提取二级质谱图,查看是否有核心岩藻糖特征性的中性丢失离子对出现,发现这两个糖肽谱图虽然含有糖特征碎片离子,但是并没有发现核心岩藻糖中性丢失的峰,所以可以判定该结果是假阳性结果。
表1、yeast糖修饰鉴定结果
实施例3:肝癌细胞hepg2和lm3
将肝癌细胞hepg2和lm3混悬于8m尿素溶液中,冰上操作超声提取蛋白质混合物。运用fasp酶切方式提取的蛋白混合物用trypsin酶进行酶切得到肽段混合物,然后通过运用hilic富集填料自制的tip小柱进行富集得到糖肽混合物;再先后使用糖苷内切酶endoh(ph5.0-6.0)和endof3(ph4.0-5.0)进行酶切,将糖肽糖链部分的外围天线全部切除,剩余产物为连接有一个最内侧n-乙酰葡糖胺glcnac的肽段和连接有一个最内侧的n-乙酰葡糖胺加一个核心岩藻糖glcnac+fuc的肽段的混合物,记为简化糖肽样本;所述glcnac记为hn;所述glcnac+fuc记为cf;连接有glcnac的肽段记为hn肽段;连接有glcnac+fuc的肽段记为cf的肽段;将样品热干脱盐后送入液质联用检测。
液质联用部分方法为:
a.液相部分:同实施例1;
b.质谱部分:同实施例1;
定性数据处理:运用proteomediscoverer2.1软件(美国,thermofisher公司)将raw文件转换为mgf格式,默认参数设置。运用perl语言编写的程序包对转换后的mgf格式文件进行谱图挑选,挑选原则为对含有糖特征碎片的信号离子进行谱图提取,进而对谱图中核心岩藻糖特征的中性丢失母离子进行筛查,若筛查出中性丢失母离子,则对候选谱图进行母离子重校正以及糖特征碎片离子去除。然后将处理得到的文件利用pfind2.8.0.1软件进行数据库检索。使用homosapiensuniprotdatabase(人)(20205个条目)作为背景数据库。用字母“j”替换所有背景数据库中符合n-糖基化特征序列(nx(s/t/c),x不为p)的“n”,目的是将“j”定义为与天冬酰胺(n)具有相同分子量的氨基酸。用pfind2.8.0.1生产对应的反库,采用正反库(target-decoy)方法进行数据库检索。该数据库检索时,设置可变修饰:蛋氨酸氧化(oxidation_met)、蛋白质n末端乙酰化(acetylation_protein_n_terminal)以及j残基的203.079da的质量改变(_glcnac_j_203.079),固定修饰设置为:半胱氨酸烷基化(carbamidomethylation_cys)。最多允许2个trypsin漏切位点,母离子误差10ppm,子离子误差15mmu,可容许1%的肽段水平假阳性率(fdr)。
定量数据处理:首先将raw文件利用proteinwizard软件中msconvert模块转换为mzxml格式,再利用r语言中的readmzxmlpackage,从mzxml文件中提取ms1完整信息(包括实际保留时间值(rt)、对应的谱图序号以及一级峰强度)ms1.txt,另一方面对搜库结果利用自编的处理程序得到简化糖肽的scannumber和m/z信息。利用此信息,结合ms1.txt文件中的详细信息,得到该糖肽的rt推测值以及理论质量数,并根据质量差和保留时间差的规则,利用已获得的hn肽段或cf的肽段的rt值和理论质量数去推测同一位点上cf的肽段或hn肽段的rt值和理论质量数;从而进一步通过rt推测值提取ms1的峰面积作为该糖肽的定量值信息。
质量差规则为hn肽段和cf的肽段的质量数相差一个核心岩藻糖的质量数;
保留时间差的规则为hn肽段和cf的肽段的保留时间差在1min内;
以肝癌的两个细胞系hepg2和lm3作为实验对象,为了验证实验的可重复性和稳定性,分别进行了三次平行实验,共计鉴定到hepg2中含cf的肽段有574条,含cf修饰的蛋白318个;含hn修饰的肽段1102条,含hn修饰蛋白的561个。lm3三次平行实验共计鉴定含cf的肽段有555条,含cf修饰的蛋白298个;含hn修饰的肽段844条,含hn修饰蛋白的453个。另外,为了验证实验数据的可重复性及稳定性,对肝癌的两个细胞系hepg2和lm3三次平行实验的定量结果分别作了相关性分析,可以看到r值均大于0.9(图5中a),结果呈现良好的稳定性和可重复性。
在两种推演方法共同鉴定到的含cf修饰的肽段核心岩藻糖占有率相关性分析中,可以看到r值近似与1(图5中b),说明两种推演方法结果一致,可信。
在hepg2和lm3中共同鉴定到含cf修饰的蛋白有103个,共同鉴定到163个含cf修饰的肽段。而只在hepg2中鉴定到的含cf修饰的蛋白有111个,只在lm3中鉴定到的含cf修饰的蛋白有117个。
图6为肝癌细胞定量数据cf-ratio特征分析图。由图可知,在两种肝癌细胞系hepg2和lm3中,核心岩藻糖的位点占有比率是有差异的,这也为探索差异修饰的核心岩藻糖修饰的蛋白质作为肝癌诊断标志物提供了可行性。
- 还没有人留言评论。精彩留言会获得点赞!