一种全基因组测序数据计算解读方法与流程
本发明涉及基因测序技术,具体涉及一种全基因组测序数据计算解读方法。
背景技术:
近年来,随着下一代测序技术(nextgenerationsequence,ngs)的广泛应用,基因测序的成本迅速下降,基因测序技术得以在更加广泛的生物、医疗、健康、刑侦、农业等等许多领域被推广应用。其中,基于ngs的全基因组测序(wholegenomesequencing,wgs)是一个非常有应用价值的分支领域,受到广泛的关注。
全基因组测序是指对一种生物个体的基因组中的全部基因进行测序,即测定其脱氧核糖核酸(deoxyribo-nucleicacid,dna)的碱基序列。基因组信息已能用于鉴定遗传疾病,查找驱使癌症发展的突变,追踪疾病的爆发。而全基因组测序可谓是基因组最为全面的研究方案和最强有力的研究工具。尽管全基因组测序常被理解为用于测定人类基因组,但实际上ngs的规模和灵活性体现于可以在任何物种上高效运用全基因组测序技术,如农业畜牧业,植物,或疾病相关微生物。
基于ngs的全基因组测序的优点在于:能够提供高分辨率、精确到逐个碱基的基因组视图;可以捕获大的变异,以及小到可能被遗漏的变异;能够鉴定潜在的致病变异,从而进行基因表达和调控机制的进一步研究;在短时间内提供大量的数据,以支持新基因组的组装等。例如,外显子组测序或靶向重测序等有侧重点的方法只分析基因组的有限部分,全基因组测序则不同,能提供整个基因组的全面视图。它是各种发现应用——如鉴定致病变异和新基因组组装——的理想选择。全基因组测序可检测单核苷酸多态性(singlenucleotidepolymorphism,snp)、插入/缺失(indel)、拷贝数变异(copynumbervariation,cnv)和大的(large)结构化变异(structuralvariants,sv)。随着技术创新,最新的基因组测序仪能够比以往更高效地开展全基因组测序。
基于ngs的全基因组测序的数据处理流程包括数据计算和数据解读两大步骤,其中的数据计算步骤完成参考基因组的预处理和原始测序数据的修剪、比对、去重等计算任务,以便数据解读时使用;数据解读步骤对数据计算处理后的数据在生物学、医学、健康保健等领域的科学含义进行分析、揭示和解释。
目前,基于ngs的全基因组测序技术在应用上存在两个方面的瓶颈:
一个瓶颈是测序数据产出能力远远大于测序数据处理能力。例如,在基于ngs的全基因组测序中比较常用的一个测序数据计算解读协议gatk流程中,基于参考基因组na12878,处理一个人的全基因组测序样本数据(大约100gb),在cpu上仅仅是进行深度覆盖的单倍型识别(haplotypecaller)这一个子步骤,就大约需要耗时10到15天,而illumina公司的hiseq4000测序仪在5个小时之内能够产出200m个读长为300碱基对(basepair,bp)的测序片段(reads)。因此,一方面,测序生成的原始数据每年3到5倍的增加速度已经远超摩尔定律,而测序数据的计算解读又是高输入/输出密集和高计算密集型任务,对测序数据进行实时的、准确的计算解读和传送变得非常困难,面临着巨大的挑战。另一方面,目前典型的测序数据计算解读方法仍然主要是依托高性能的中央处理器(centralprocessingunit,简称cpu),运用基于多线程技术的软件进行处理。但是,在保证准确性的前提下,它能获得的计算解读加速性能仍然无法满足上述挑战的需求。所以,这种方法已经缺乏持续性。
另一个瓶颈是测序数据解读的深度、广度无法满足科研人员的需求,与此同时其可读性又无法满足普通大众的需求。目前测序数据解读的典型方法是基于一个参考基因组,然而,当前使用的参考基因组本身就是基于有限的样本,既不足以代表整个相关物种的多样性,又不完备,因此在数据计算解读时会导致偏差,而且缺乏与其它生物、医学信息的广泛地、深度交叉分析,难以满足专业科研人员深入研究的需求。此外,测序数据解读还基本停留在专业领域,面向非专业的大众,又缺乏可读性,即缺乏对测序数据直接的生物意义和间接的健康影响的通俗易懂、形式多样的解读。
目前,信息处理领域常见的处理器类型有中央处理器(centralprocessingunit,简称cpu)、现场可编程门阵列(fieldprogrammablegatearray,简称fpga)、图形处理器(graphicsprocessingunit,简称gpu)和数字信号处理器(digitalsignalprocessor,简称dsp)。高性能cpu通常都包括多个处理器核(processorcore),从硬件上支持多线程,但是其设计目标还是面向通用应用程序,而相对于特殊的计算,通用应用程序的并行性较小,需要较复杂的控制和较低的性能目标。因此,cpu片上的硬件资源主要还是用于实现复杂的控制而不是计算,没有为特殊功能包含专门的硬件,能够支持的计算并行度不高。fpga是一种半定制电路,优点有:基于fpga进行系统开发,设计周期短、开发费用低;功耗低;生产后可重新修改配置,设计灵活性高,设计风险小。缺点是:实现同样的功能,fpga一般来说比专用集成电路(applicationspecificintegratedcircuit,asic)的速度要慢,比asic电路面积要大。随着技术的发展和演进,fpga向更高密度、更大容量、更低功耗和集成更多硬核知识产权(intellectualproperty,ip)的方向发展,fpga的缺点在缩小,而优点在放大。相比于cpu,fpga可以用硬件描述语言来定制实现、修改和增加并行计算。gpu最初是一种专门用于图像处理的微处理器,能够从硬件上支持纹理映射和多边形着色等图形计算基本任务。由于图形学计算涉及一些通用数学计算,比如矩阵和向量运算,而gpu拥有高度并行化的架构,因此,随着相关软硬件技术的发展,gpu计算技术日益兴起,即gpu不再局限于图形处理,还被开发用于线性代数、信号处理、数值仿真等并行计算,可以提供数十倍乃至于上百倍于cpu的性能。但是目前的gpu存在2个问题:一是,受限于gpu的硬件结构特性,很多并行算法不能在gpu上有效地执行;二是,gpu运行中会产生大量热量,能耗较高。dsp是一种用数字方法对各种信号进行快速分析、变换、滤波、检测、调制、解调等运算处理的微处理器。为此,dsp在芯片内部结构上做了特殊的优化,比如硬件实现高速、高精度的乘法等。随着数字时代的到来,dsp广泛应用于智能设备、资源勘探、数字控制、生物医学、航天航空等各个领域,具有功耗低、精度高、可进行二维与多维处理等特点。综上所述,以上四种计算器件各有特点,又各有局限性。
针对前述基于ngs的全基因组测序技术应用发展存在的两个方面的瓶颈,如何利用上述处理器来实现海量测序数据的快速实时、精准深入、通俗易懂、形式多样的计算解读,则已经成为一项亟待解决的关键技术问题。
技术实现要素:
本发明要解决的技术问题:针对现有技术的上述问题,提供一种快速实时、精准深入、通俗易懂、形式多样的全基因组测序数据计算解读方法。
为了解决上述技术问题,本发明采用的技术方案为:
一种全基因组测序数据计算解读方法,实施步骤包括:
1)输入用于全基因组测序的参考全基因组数据、原始测序样本数据;基于cpu对原始测序样本数据进行预处理得到测序样本可靠数据;如果参考全基因组数据需要预处理,则基于cpu对参考全基因组数据进行预处理得到作为比对对象的带索引的参考全基因组数据,且cpu调用通过fpga硬件实现的索引生成器对生成索引进行硬件加速;如果参考全基因组数据不需要预处理,则直接将原始的带索引的参考全基因组数据作为比对对象;
2)由cpu调用fpga加速将测序样本可靠数据和带索引的参考全基因组数据进行比对,得到具有重复标志、带索引的比对结果;
3)由cpu调用fpga和gpu加速对测序样本可靠数据进行基因组重新装配,并对具有重复标志、带索引的比对结果进行变异识别发现;
4)由cpu调用gpu和dsp加速对重新装配后的测序样本可靠数据、具有重复标志、带索引的比对结果的变异识别发现结果进行可视化处理,并基于可视化处理结果由cpu调用fpga上硬件实现的深度学习模型负责并行执行全基因组及变异功能分析和挖掘。
优选地,步骤1)中基于cpu对原始测序样本数据进行预处理得到测序样本可靠数据具体是指进行数据质量控制,所述数据质量控制包括修剪全基因组测序原始样本数据,以及移除reads上的接头序列、低质量序列、污染物、人造物,从而得到测序样本可靠数据。
优选地,步骤1)中基于cpu对原始测序样本数据进行预处理、基于cpu对参考全基因组数据进行预处理是在cpu上采用不同的线程并发执行。
优选地,步骤2)的详细步骤包括:
2.1)读取测序样本可靠数据以及作为比对对象的带索引的参考全基因组数据;
2.2)根据参考全基因组数据的索引,由cpu调用通过fpga硬件实现的比对器将测序样本可靠数据和带索引的参考全基因组数据进行比对;
2.3)根据比对结果,cpu调用通过fpga硬件实现的排序器对测序样本可靠数据进行排序和标重;
2.4)根据比对及排序和标重的结果,对测序可靠样本数据进行数据清洗,所述数据清洗包括进行indel重新比对、碱基的重新校准和数据合并,进行indel重新比对是由cpu调用通过fpga上硬件实现的比对器完成;
2.5)输出有重复标志的、带索引的比对结果信息。
优选地,步骤3)中对测序样本可靠数据进行基因组重新装配的详细步骤包括:
3.1a)读取测序样本可靠数据;
3.2a)通过cpu调用fpga硬件实现的装配器将测序样本可靠数据重新装配成基因组,得到包含测序样本可靠数据和重新装配后得到的新基因组之间的映射关系;
3.3a)输出重新装配的基因组。
优选地,步骤3)中对具有重复标志、带索引的比对结果进行变异识别发现的详细步骤包括:
3.1b)读取具有重复标志、带索引的比对结果;
3.2b)判断比对结果是否双端一致,如果是双端不一致,则跳转执行步骤3.3b);否则,跳转执行步骤3.5b);
3.3b)由cpu调用gpu上编程实现的识别器针对具有重复标志、带索引的比对结果进行大的结构化变异识别,所述大的结构化变异影响的基因序列长度在1000到3百万个碱基之间;
3.4b)输出识别出的大结构化变异结果信息,跳转执行步骤4);
3.5b)进行变异识别发现;
3.6b)输出识别出的变异结果信息。
优选地,步骤3.5b)基于cpu进行变异识别发现时,包括基于cpu进行基因拷贝数变异识别、进行其它变异识别,所述其它变异识别包括snp和小indel识别;步骤3.6b)输出识别出的变异结果信息包括基因拷贝数变异识别的结果信息、snp和小indel识别的结果信息。
优选地,所述基于cpu和fpga进行其它变异识别具体是指由cpu调用fpga硬件实现的识别器进行。
优选地,步骤4)的详细步骤包括:
4.1)读取重新装配后的测序样本可靠数据、具有重复标志、带索引的比对结果的变异识别发现结果;
4.2)将重新装配后的测序样本可靠数据、具有重复标志、带索引的比对结果的变异识别发现结果进行可视化处理,且进行可视化处理时,由cpu调用gpu完成可视化处理的视频、动画和显示任务,由cpu调用dsp完成可视化处理的图形、图像和音频任务;
4.3)基于可视化处理结果进行全基因组及变异功能分析和挖掘;
4.4)输出分析数据和深度解读报告。
优选地,步骤4.3)基于可视化处理结果进行全基因组及变异功能分析和挖掘具体是指由cpu调用fpga上硬件实现的深度学习模型负责并行执行分析和挖掘,且由cpu调用gpu完成分析和挖掘相关的视频、动画和显示任务、由cpu调用dsp完成分析和挖掘相关的图形、图像和音频任务。
本发明全基因组测序数据计算解读方法具有下述优点:
1、对于全基因组测序数据计算解读流程中的每个耗时瓶颈,基于任务本身的算法或模型并行性,结合cpu、fpga、gpu和dsp这四种处理器的特点,分别进行了有针对性的并行加速,提高了全基因组测序数据计算解读的实时性。
2、对于全基因组测序数据计算解读流程中的全基因组和变异功能分析与挖掘,基于任务本身的目标,结合cpu、fpga、gpu和dsp这四种处理器的特点,引入了深度学习模型,加快和丰富了深度学习源数据的处理,提高了全基因组测序数据计算解读的深度和广度。
3、对于全基因组测序数据计算解读流程中的数据可视化,结合cpu、gpu和dsp这三种处理器的特点,配合完成可视化处理,提高了全基因组测序数据可视化的实时性,丰富了全基因组测序数据可视化的多样性。
附图说明
图1为本发明实施例全基因组测序数据计算解读的总流程示意图。
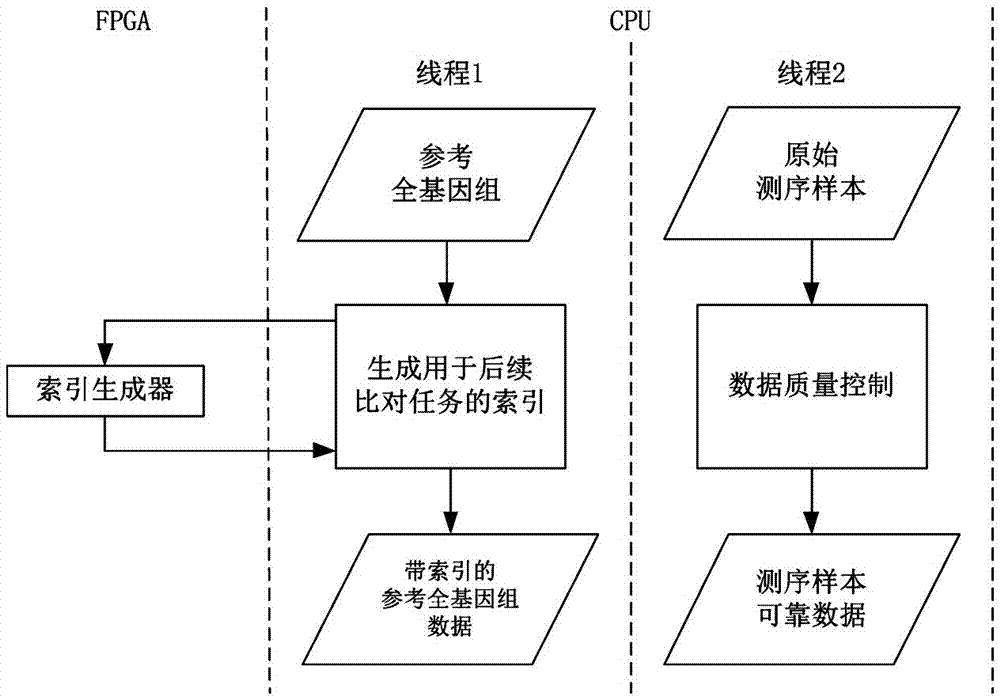
图2为本发明实施例全基因组测序数据计算解读的预处理流程示意图。
图3为本发明实施例全基因组测序数据计算解读的数据比对流程示意图。
图4为本发明实施例全基因组测序数据计算解读的变异识别和发现流程示意图。
图5为本发明实施例全基因组测序数据计算解读的数据可视化和全基因组及变异功能的分析挖掘流程示意图。
具体实施方式
如图1所示,本实施例全基因组测序数据计算解读方法的实施步骤包括:
1)输入用于全基因组测序的参考全基因组数据、原始测序样本数据;基于cpu对原始测序样本数据进行预处理得到测序样本可靠数据;如果参考全基因组数据需要预处理,则基于cpu对参考全基因组数据进行预处理得到作为比对对象的带索引的参考全基因组数据,且cpu调用通过fpga硬件实现的索引生成器对生成索引进行硬件加速;如果参考全基因组数据不需要预处理,则直接将原始的带索引的参考全基因组数据作为比对对象;这个步骤需要使用cpu和fpga这两种处理器;
2)由cpu调用fpga加速将测序样本可靠数据和带索引的参考全基因组数据进行比对(alignment),得到具有重复标志、带索引的比对结果;这个步骤需要使用cpu和fpga这两种处理器;
3)由cpu调用fpga和gpu加速对测序样本可靠数据进行基因组重新装配(denovoassembly),并对具有重复标志、带索引的比对结果进行变异识别(变异识别,variantcalling,vc)发现(变异发现,variantdiscovery);这个步骤需要使用cpu、fpga和gpu三种处理器;
4)由cpu调用gpu和dsp加速对重新装配后的测序样本可靠数据、具有重复标志、带索引的比对结果的变异识别发现结果进行可视化处理,并基于可视化处理结果由cpu调用fpga上硬件实现的深度学习(deeplearning,dl)模型负责并行执行全基因组及变异功能分析和挖掘。这个步骤需要使用cpu、fpga、gpu和dsp这四种处理器。
参见图1可知,步骤1)和2)完成全基因组测序数据的计算任务;步骤3)和4)完成全基因组测序数据的解读任务。以下详细步骤描述中,如果未加特殊说明,缺省使用的是cpu。
本实施例中,步骤1)中基于cpu对原始测序样本数据进行预处理得到测序样本可靠数据具体是指进行数据质量控制,所述数据质量控制包括修剪全基因组测序原始样本数据,以及移除reads上的接头序列(theadaptersequences)、低质量序列(low-qualitysequences)、污染物(contaminants)、人造物(artifacts),从而得到测序样本可靠数据(cleandatas)。
如图2所示,本实施例中步骤1)中基于cpu对原始测序样本数据进行预处理、基于cpu对参考全基因组数据进行预处理是在cpu上采用不同的线程(参见图2,本实施例中以线程i和线程ii为例进行说明)并发执行。步骤1)包括2个并发执行的子任务:参考全基因组的预处理和全基因组测序原始样本数据的预处理。
本实施例中,可根据需要做参考全基因组的预处理:在实际的全基因组测序数据计算解读流程中,若想最大化利用已知的全基因组研究领域的成果,就做参考全基因组的预处理;若没有参考全基因组数据或想发现新的基因组和变异,就不做参考全基因组的预处理。在为参考全基因组数据生成用于后续比对任务的索引时,cpu负责索引生成的流程控制和部分计算任务,fpga上硬件实现的索引生成器负责并行加速完成其中的计算密集任务,cpu和fpga之间有数据和指令交互。只使用cpu时,这一步骤是整个全基因组测序数据计算解读流程中的耗时瓶颈之一。虽然一段时间内,特定的参考全基因组数据相对固定,可以生成索引一次,再在同类应用中反复使用,但是,一旦参考全基因组数据有了更新,必须重新生成新索引。
如图3所示,本实施例中步骤2)的详细步骤包括:
2.1)读取测序样本可靠数据以及作为比对对象的带索引的参考全基因组数据;
2.2)根据参考全基因组数据的索引,由cpu调用通过fpga硬件实现的比对器1将测序样本可靠数据和带索引的参考全基因组数据进行比对;只使用cpu时,这一步骤是整个全基因组测序数据计算解读流程中的耗时瓶颈之一。由cpu调用通过fpga硬件实现的比对器1将测序样本可靠数据和带索引的参考全基因组数据进行比对,能够利用fpga实现比对加速,cpu负责数据比对的流程控制和部分计算任务,fpga上硬件实现的比对器1负责并行加速完成其中的计算密集任务,cpu和fpga之间有数据和指令交互。
2.3)根据比对结果,cpu调用通过fpga硬件实现的排序器对测序样本可靠数据进行排序(sort)和标重(markduplicate);其中,cpu负责数据排序和标重的流程控制和部分计算任务,fpga上硬件实现的排序器负责并行加速完成其中的计算密集任务,cpu和fpga之间有数据和指令交互。只使用cpu时,这一步骤是整个全基因组测序数据计算解读流程中的耗时瓶颈之一;
2.4)根据比对及排序和标重的结果,对测序可靠样本数据进行数据清洗(dataclean),数据清洗包括进行indel重新比对(realignment)、碱基的重新校准(baserecalibration)和数据合并(merge)等,进行indel重新比对是由cpu调用通过fpga上硬件实现的比对器2完成;其中,cpu负责数据比对的流程控制和部分计算任务,fpga上硬件实现的比对器2负责并行加速完成其中的计算密集任务,cpu和fpga之间有数据和指令交互。只使用cpu时,这一步骤是整个全基因组测序数据计算解读流程中的耗时瓶颈之一。
2.5)输出有重复标志的、带索引的比对结果信息。例如,常用的块压缩的(blockcompression)测序比对/映射格式(sequencealignment/mapformat,sam)的bam文件。
上述步骤2.3)和2.4)中fpga上硬件实现的比对器,可以是同一个硬件实现(以提高fpga硬件的复用度),此外也可以采用不同的硬件实现。
本实施例中,步骤3)由cpu调用fpga和gpu加速对测序样本可靠数据进行基因组重新装配(denovoassembly),并对具有重复标志、带索引的比对结果进行变异识别(变异识别,variantcalling,vc)发现(变异发现,variantdiscovery),该步骤包括2个并发执行的子任务:上述全基因组测序可靠样本数据重新装配成基因组;基于上述有重复标志的、带索引的比对结果信息的变异识别发现。根据需要,在实际的全基因组测序数据计算解读流程中,对于这2个子任务,若想最大化利用已知的全基因组研究领域的成果并且有参考全基因组数据,就做基于上述有重复标志的、带索引的比对结果信息的变异识别发现;若没有参考全基因组数据或想发现新的基因组和变异,就做上述全基因组测序可靠样本数据重新装配成基因组;若想兼顾,就都做。
如图4所示,步骤3)中对测序样本可靠数据进行基因组重新装配的详细步骤包括:
3.1a)读取测序样本可靠数据;
3.2a)通过cpu调用fpga硬件实现的装配器将测序样本可靠数据重新装配成基因组,得到包含测序样本可靠数据和重新装配后得到的新基因组之间的映射关系;其中,cpu负责基因组重新装配的流程控制和部分计算任务,fpga上硬件实现的装配器负责并行加速完成其中的计算密集任务,cpu和fpga之间有数据和指令交互。只使用cpu时,这一步骤是整个全基因组测序数据计算解读流程中的耗时瓶颈之一;
3.3a)输出重新装配的基因组。
如图4所示,步骤3)中对具有重复标志、带索引的比对结果进行变异识别发现的详细步骤包括:
3.1b)读取具有重复标志、带索引的比对结果;
3.2b)判断比对结果是否双端一致(concordantpairs),如果是双端不一致(discordantpairs),则跳转执行步骤3.3b);否则,跳转执行步骤3.5b);
3.3b)由cpu调用gpu上编程实现的识别器2针对具有重复标志、带索引的比对结果进行大sv识别,大sv影响的基因序列长度在1000到3百万个碱基之间,例如:非整倍体(aneuploidies)、标记染色体(markerchromosome)、总重排(grossrearrangements)、染色体大小变异(variationinchromosomesize)等;其中,cpu负责大结构化变异识别的流程控制和部分计算任务,gpu上编程实现的识别器2负责并行加速完成其中的计算密集任务,cpu和gpu之间有数据和指令交互。只使用cpu时,这一步骤是整个全基因组测序数据计算解读流程中的耗时瓶颈之一。
3.4b)输出识别出的大结构化变异结果信息,跳转执行步骤4);
3.5b)进行变异识别发现;
3.6b)输出识别出的变异结果信息。
本实施例中,步骤3.5b)基于cpu进行变异识别发现时,包括基于cpu进行基因cnv识别、进行其它变异识别,所述其它变异识别包括snp和小indel识别;步骤3.6b)输出识别出的变异结果信息包括基因拷贝数变异识别的结果信息、snp和小indel识别的结果信息。cnv是由基因组发生重排而导致的,一般指长度为1kb以上的基因组大片段的拷贝数增加或者减少,主要表现为亚显微水平的缺失和重复。cnv影响的基因组片段大小比snp和小indel要大,而且cnv位点的突变率远高于snp,是人类疾病的重要致病因素之一,因此单独进行识别。其它变异识别,包括snp和小indel识别。其中,cpu负责变异识别的流程控制和部分计算任务,fpga上硬件实现的识别器1负责并行加速完成其中的计算密集任务,cpu和fpga之间有数据和指令交互。只使用cpu时,这一步骤是整个全基因组测序数据计算解读流程中的耗时瓶颈之一。
本实施例中,基于cpu和fpga进行其它变异识别具体是指由cpu调用fpga硬件实现的识别器1进行。
如图5所示,步骤4)的详细步骤包括:
4.1)读取重新装配后的测序样本可靠数据、具有重复标志、带索引的比对结果的变异识别发现结果;
4.2)将重新装配后的测序样本可靠数据、具有重复标志、带索引的比对结果的变异识别发现结果进行可视化处理,且进行可视化处理时,由cpu调用gpu完成可视化处理的视频、动画和显示任务,由cpu调用dsp完成可视化处理的图形、图像和音频任务;通过进行可视化处理,能够以各种科学、直观、生动的方式表现数据的含义。其中,cpu负责可视化的流程控制和部分计算任务;gpu上编程处理视频、动画和显示等任务,cpu和gpu之间有数据和指令交互;dsp上编程处理图形、图像和音频等任务,cpu和dsp之间有数据和指令交互。只使用cpu时,这一步骤是整个全基因组测序数据计算解读流程中的耗时瓶颈之一,加入gpu和dsp,它们和cpu相互配合,能够并行加速完成多媒体处理任务。
4.3)基于可视化处理结果进行全基因组及变异功能分析和挖掘,即基于上述分析数据,进一步深入分析相关的全基因组和变异的功能,例如:变异鉴定、分类研究;基因组和变异与物种特性之间的关联性研究;基因组和变异与已知疾病之间的关联性研究;并扩大外延,在已知的知识之外,再挖掘未知的关联。例如:发现全新(denovo)变异,分析、挖掘、预测和鉴定它们的作用等;
4.4)输出分析数据和深度解读报告。
本实施例中,步骤4.3)基于可视化处理结果进行全基因组及变异功能分析和挖掘具体是指由cpu调用fpga上硬件实现的深度学习模型负责并行执行,且由cpu调用gpu完成分析和挖掘相关的视频、动画和显示任务、由cpu调用dsp完成分析和挖掘相关的图形、图像和音频任务。其中,cpu负责分析和挖掘的流程控制和部分计算任务;fpga上硬件实现的深度学习模型负责并行执行分析和挖掘,cpu和fpga之间有数据和指令交互。使用深度学习方法进行分析和挖掘,可以支持基于大数据的统计学模型,能够实现更加准确的分析和挖掘;gpu上编程处理分析挖掘相关的视频、动画和显示等任务,cpu和gpu之间有数据和指令交互;dsp上编程处理和分析挖掘相关的图形、图像和音频等任务,cpu和dsp之间有数据和指令交互。只使用cpu时,这一步骤是整个全基因组测序数据计算解读流程中的耗时瓶颈之一,加入fpga、gpu和dsp,它们和cpu相互配合,能够并行加速完成深度学习及相关多媒体处理任务;
综上所述,本实施例的全基因组测序数据计算解读方法能够满足测序数据计算解读的快速实时、精准深入、通俗易懂、形式多样的要求,为全基因组测序技术的应用推广助力。
以上所述仅是本发明的优选实施方式,本发明的保护范围并不仅局限于上述实施例,凡属于本发明思路下的技术方案均属于本发明的保护范围。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理前提下的若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!