基于信息论基因转录本组装与量化方法及系统与流程
本发明涉及计算生物学技术领域,尤其涉及一种基于信息论基因转录本组装与量化方法及系统。
背景技术:
随着下一代基因测序技术的发展,基因生物学研究迫切需要有效的量化方法,来对高通量rna测序的内在基因调控与转录情况进行解析。在rna层面,转录本的识别与丰度估计是评估转录功能差异性的重要方法,在新一代测序研究中能揭示疾病潜在的机理,发现新的生物结论。转录本组装是从大规模测序读段中有结构的恢复基因所表达出来的转录本变体。峰度估计是对发现的转录本的表达水平进行量化估计。然而,要完成这两项任务,仅有的数据是从转录本片段中推测完整的测序信息。从有限的观测中获取完整的数据解析本质上是一个病态的数学问题。由于缺失信息的存在,在得到对结果中会出现显著的不确定性。
传统的转录本发现与丰度量化方法采用的是基于多种不同考虑建立的参数统计方法,比如概率生成模型护着是线性回归模型。尽管他们的数学表示存在很大的差别,内在的数学概念仍然是同属于相似的数据拟合类别。从转录本到rna测序的读段这个测序过程中,会由于信息缺失和数据模糊引入显著的不确定水平。例如,转录本元素的不确定性,rna测序读段映射的多样性,读段在转录本上分布的分均一性等,这些都是很难控制的不确定元素。当数据拟合任务遇到众多不确定性时,在最终估计结果中会引入无法避免的偏差。
许多数据拟合方法依赖于外部的信息来减少数据的不确定性,可能需要部分或者全部的基因组注释来指导转录本组装。但是目前相关技术中的方法的精度有限,需要进一步提升。而且尽管这些方法的数学基础是非常相似的,但是被这些不同方法发现的转录本存在着较大的差异。因此,仍然需要更精确与通用的无需基因注释的转录本推断与量化方法。
技术实现要素:
本发明旨在至少在一定程度上解决相关技术中的技术问题之一。为此,本发明的一方面目的在于提出一种能够不依赖于外部的基因位置标记,基因组装准确性显著提高,提升测序精度的基于信息论基因转录本组装与量化方法。
本发明另一方面目的在于提出一种基于信息论基因转录本组装与量化系统。
为达到上述目的,本发明一方面的实施例提出了一种基于信息论基因转录本组装与量化方法,包括以下步骤:将测序的读段与参照的基因组对齐,并根据测序的读段与参照的基因组对齐结果对初始的基因与转录本的开始位置与终止位置进行预测;在预测完之后,建立有向图以模拟可能的转录本,得到候选转录本集合;根据最大化信息传输容量的方式对候选转录本集合进行转录本预测与峰度估计。
根据本发明实施例的基于信息论基因转录本组装与量化方法,通过对齐测序的读段与参照的基因组,根据结果预测初始的基因与转录本的开始位置与终止位置,并建立候选转录本集合,根据最大化信息传输容量的方式对候选转录本集合进行转录本预测与峰度估计。该方法不依赖于外部的基因位置标记,基因组装准确性显著提高,提升测序精度。
在一些示例中,所述对初始的基因进行预测,包括:子外显子发现、基因边界预测和基因结构预测。
在一些示例中,所述在预测完之后,建立有向图以模拟可能的转录本,得到候选转录本集合,包括:根据所述有向图翻译基因剪切变体,以模拟可能的转录本,得到候选转录本集合。
在一些示例中,所述根据最大化信息传输容量的方式对候选转录本集合进行转录本预测与峰度估计,包括:根据目标函数对候选转录本集合进行转录本预测与峰度估计,其中,所述目标函数为:
maxi(t;r|θ)+λl(θ;r),
其中,l(θ;r)=logp(r|θ)为似然项,λ平衡了不确定性与似然的相对重要性。
在一些示例中,所述的基于信息论基因转录本组装与量化方法,其特征在于,
其中,
其中,当所述目标函数达到最大值时前向选择终止。
本发明的另一方面的实施例提出了一种基于信息论基因转录本组装与量化系统,包括:初始的基因预测模块,用于将测序的读段与参照的基因组对齐,并根据测序的读段与参照的基因组对齐结果对初始的基因与转录本的开始位置与终止位置进行预测;候选转录本集合获取模块,用于在预测完之后,建立有向图以模拟可能的转录本,得到候选转录本集合;转录本预测与峰度估计模块,用于根据最大化信息传输容量的方式对候选转录本集合进行转录本预测与峰度估计。
根据本发明实施例的基于信息论基因转录本组装与量化系统,通过对齐测序的读段与参照的基因组,根据结果预测初始的基因与转录本的开始位置与终止位置,并建立候选转录本集合,根据最大化信息传输容量的方式对候选转录本集合进行转录本预测与峰度估计。该系统不依赖于外部的基因位置标记,基因组装准确性显著提高,提升测序精度。
本发明附加的方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
图1是根据本发明实施例的基于信息论基因转录本组装与量化方法的流程图;
图2是根据本发明一个实施例的基于信息论基因转录本组装与量化方法技术实施方案的流程图;和
图3是本发明一个实施例的基于信息论基因转录本组装与量化系统的结构示意图。
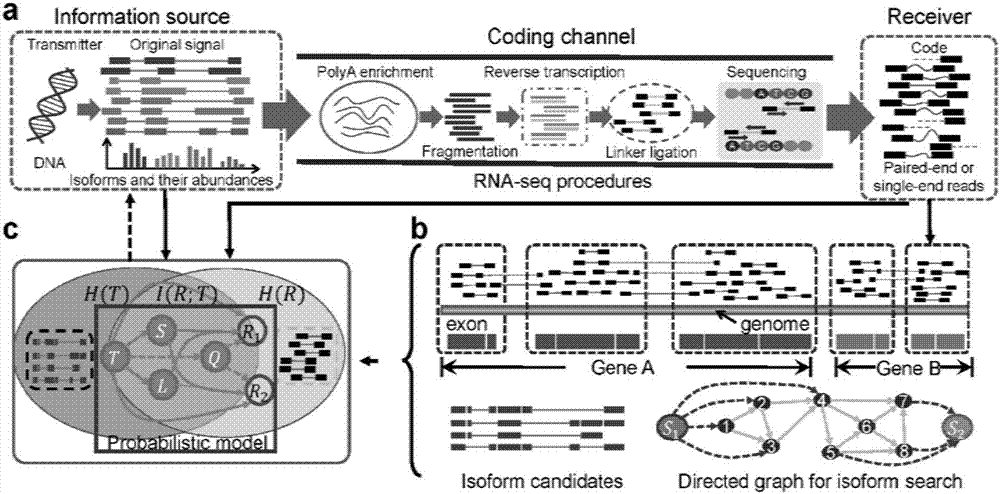
图2中,a)为本方法所研究的问题,转录本到测序读段之间的测序过程通过信道传输来进行模拟,转录本作为信息源,测序读段作为得到的接收到的编码信号;b)为候选基因预测与候选基因重建;图中两个基因(a和b)定位在基因组上,由读段的分布来决定;在基因a中,8个子外显子被识别出来,用于构建有向图的节点;一对初始与终止节点(s1,s2)加在图中用以标注来时与终止节点;c)为信息传输模型;h(t)和h(r)代表着转录本与读段的熵,i(t;r)是互信息,用来衡量转录本与读段之间的信息共享;概率图模型被引入来解析从转录本(t)到rna测序读段(r)的过程;r1,r2代表一组读段。在图中,s与l代表转录本的开始位置与片段长度,q代表读段匹配的质量。
具体实施方式
下面详细描述本发明的实施例,所述实施例的示例在附图中示出,其中自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。
图1为根据本发明实施例的基于信息论基因转录本组装与量化方法的流程图及图2根据本发明一个实施例的基于信息论基因转录本组装与量化方法技术实施方案的流程图。结合图1和图2,该方法包括以下步骤:
s1,将测序的读段与参照的基因组对齐,并根据测序的读段与参照的基因组对齐结果对初始的基因与转录本的开始位置与终止位置进行预测
首先,基因预测通常包括三个部分:子外显子发现,基因边缘寻找与转录本起止位置预测。在具体示例中,读段首先通过tophat2与基因组进行匹配。读段中检测到的链接被认为是可能的剪切点。有较少读段支持的剪切点被排除在外来降低组装错误。两种表达的片段被推断为子外显子:在相邻的3端与5端区域以及在5端与3端的区域。
进一步地,在组装好子外显子之后,本方法决定基因的边界,将子外显子分配到不同的基因位置。基因位置首先通过他们方向信息决定。如果读段中的链接tophat2被标记为不同的方向,那就应该属于不同的基因。在局部基因区域,使用高质量的子外显子来估计子外显子长度的概率分布,特别长的子外显子被认为是可能横跨两个基因的片段。在这些具有可疑的片段中,本方法用具有明显不连续的读段分布作为基因边界。
s2,在预测完之后,建立有向图以模拟可能的转录本,得到候选转录本集合。
在具体示例中,在预测基因结构之后,通过构建有向图来翻译基因剪切变体。如图2所示,节点代表通过组装的子外显子,两个子外显子如果是相接的或者是横跨两个读段的,则对应的节点通过有向边进行连接。一堆起止点添加到图中,来作为推断的转录本可能的起止位置。所有可能的转录本候选可以通过在构建图中进行路径搜索来进行枚举。为了降低候选转录本的集合,本方法用有向图中的流来进行转录本选择。具体来说,边上的流定义为横跨两个节点之间的读段数目。每条路径通过两个指标来进行衡量:
s3,根据最大化信息传输容量的方式对候选转录本集合进行转录本预测与峰度估计。
首先,介绍一下在数学上,信道容量定义为:
其中i(r;t)是转录本与测序读段之间的互信息。t={t1,t2,…,tk}是候选的转录本集合,r={r1,r2,…,rn}是观测到的测序读段集合,p(t)是转录本的生成概率,与丰度值有关,定义θk=p(t=tk)并且θ={θ1,θ2…,θk}.可以将此概率很容易转换为正式估计量,如fpkm。
在一些示例中,信道容量定义了可以传输通过信道的最大信息量,其目标函数定义为关于参数集θ的函数:
也就是说,互信息可以解释为在观测到rna测序读段之后对于转录本的不确定度的减少。
在具体示例中,在不确定性建模以外,另一项目标是读段生成的概率p(r|θ)。这一项解释了生成测序读段的概率以及估计的结果如何与读段数据匹配。从而整体的目标函数为:
maxi(t;r|θ)+λl(θ;r),
其中,l(θ;r)=logp(r|θ)为似然项。λ平衡了不确定性与似然的相对重要性。整个模型通过expectation-maximization(em)算法求解。
在具体示例中,还要对转录本集合前向选择进一步提炼,具体包括:
当目标函数达到最大值时前向选择终止。
根据本发明实施例的基于信息论基因转录本组装与量化方法,通过对齐测序的读段与参照的基因组,根据结果预测初始的基因与转录本的开始位置与终止位置,并建立候选转录本集合,根据最大化信息传输容量的方式对候选转录本集合进行转录本预测与峰度估计。该方法不依赖于外部的基因位置标记,基因组装准确性显著提高,提升测序精度。
另外,本发明的实施例公开了基于信息论基因转录本组装与量化系统,如图3所示,为是本发明一个实施例的基于信息论基因转录本组装与量化系统的结构示意图,该基于信息论基因转录本组装与量化系统10包括:初始的基因预测模块101,候选转录本集合获取模块102和转录本预测与峰度估计模块103。
其中,初始的基因预测模块101,用于将测序的读段与参照的基因组对齐,并根据测序的读段与参照的基因组对齐结果对初始的基因与转录本的开始位置与终止位置进行预测
候选转录本集合获取模块102,用于在预测完之后,建立有向图以模拟可能的转录本,得到候选转录本集合。
转录本预测与峰度估计模块103,用于根据最大化信息传输容量的方式对候选转录本集合进行转录本预测与峰度估计。
需要说明的是,前述对基于信息论基因转录本组装与量化方法实施例的解释说明也适用于该基于信息论基因转录本组装与量化的系统,此处不再赘述。
根据本发明实施例的基于信息论基因转录本组装与量化系统,通过对齐测序的读段与参照的基因组,根据结果预测初始的基因与转录本的开始位置与终止位置,并建立候选转录本集合,根据最大化信息传输容量的方式对候选转录本集合进行转录本预测与峰度估计。该系统不依赖于外部的基因位置标记,基因组装准确性显著提高,提升测序精度。
在本发明的描述中,需要理解的是,术语“中心”、“纵向”、“横向”、“长度”、“宽度”、“厚度”、“上”、“下”、“前”、“后”、“左”、“右”、“竖直”、“水平”、“顶”、“底”“内”、“外”、“顺时针”、“逆时针”、“轴向”、“径向”、“周向”等指示的方位或位置关系为基于附图所示的方位或位置关系,仅是为了便于描述本发明和简化描述,而不是指示或暗示所指的装置或元件必须具有特定的方位、以特定的方位构造和操作,因此不能理解为对本发明的限制。
此外,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括至少一个该特征。在本发明的描述中,“多个”的含义是至少两个,例如两个,三个等,除非另有明确具体的限定。
在本发明中,除非另有明确的规定和限定,术语“安装”、“相连”、“连接”、“固定”等术语应做广义理解,例如,可以是固定连接,也可以是可拆卸连接,或成一体;可以是机械连接,也可以是电连接;可以是直接相连,也可以通过中间媒介间接相连,可以是两个元件内部的连通或两个元件的相互作用关系,除非另有明确的限定。对于本领域的普通技术人员而言,可以根据具体情况理解上述术语在本发明中的具体含义。
在本发明中,除非另有明确的规定和限定,第一特征在第二特征“上”或“下”可以是第一和第二特征直接接触,或第一和第二特征通过中间媒介间接接触。而且,第一特征在第二特征“之上”、“上方”和“上面”可是第一特征在第二特征正上方或斜上方,或仅仅表示第一特征水平高度高于第二特征。第一特征在第二特征“之下”、“下方”和“下面”可以是第一特征在第二特征正下方或斜下方,或仅仅表示第一特征水平高度小于第二特征。
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
- 还没有人留言评论。精彩留言会获得点赞!