长效药物组合物的制作方法
相关专利和专利申请的交叉引用这是一个专利合作条约申请,并且要求于2014年9月26日提交的美国临时专利申请no.62/055,779的权益,该美国临时专利申请的全部内容通过引用并入本文。本发明涉及长效肠胃外(lap)制剂以及使用该制剂治疗人类免疫缺陷病毒(hiv)感染和获得性免疫缺陷综合征(aids)的方法。
背景技术:
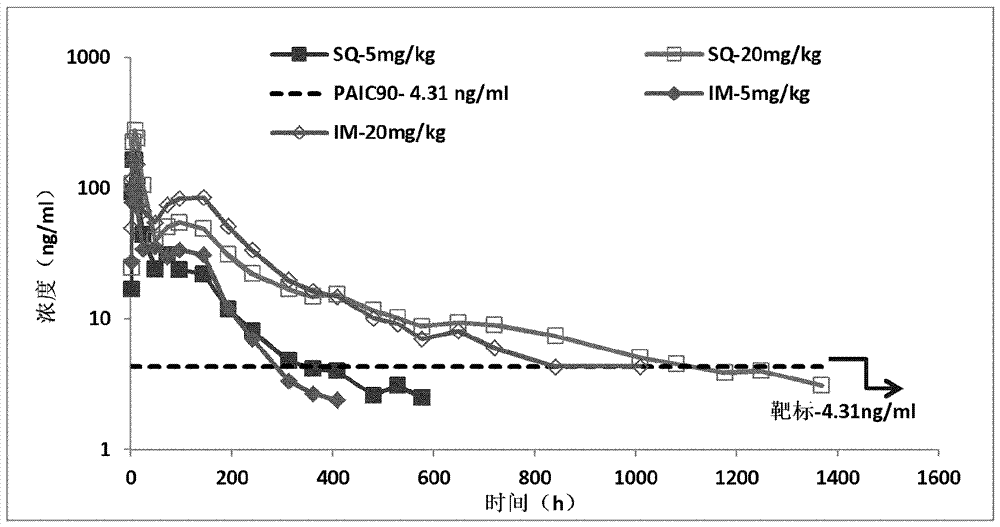
:目前,用抗逆转录病毒药物长期抑制病毒复制是治疗hiv-1感染的唯一选择。到目前为止,已经显示许多批准的药物大大增加了患者的存活。然而,被称为高活性抗逆转录病毒治疗(haart)的治疗方案通常是复杂的,因为必须向患者施用不同药物的组合以避免耐药性hiv-1变体的快速出现。此类方案通常需要频繁施用高剂量的多种药物以维持有效的药物血浆水平。因此,规定的治疗可能需要摄取多种和/或大剂量形式,这可能导致患者依从性降低,进而导致药物效力降低和hiv的多种药物抗性菌株的发展。因此,尽管haart对患者存活具有积极影响,但药物有效性和耐药性问题仍然可能发生,有时伴有致命的后果。多药耐药(mdr)hiv-1分离株的出现具有严重的临床后果,并且必须用新的药物方案(称为补救治疗)抑制。目前的指南建议补救治疗包括至少两种,优选三种完全活性药物。通常,一线治疗组合三至四种靶向病毒酶逆转录酶(rt)和蛋白酶(pr)的药物。补救治疗的一个选择是施用对耐药分离株保持活性的来自相同机制类型的药物的不同组合。然而,该方法的选择通常是有限的,因为耐药突变常常赋予对同一类型的不同药物的广泛交叉耐药性。随着融合、进入和整合酶(in)抑制剂的开发,另外的治疗策略最近变得可用。然而,已经有报道在体外和体内对所有三种新药物类型的耐药性。因此,对hiv-1感染患者的成功治疗(其缓解依从性问题并且对耐药菌株有效)是持续需要的。发明概述本发明通过将某些hiv抑制剂化合物,包括第一化合物、第二化合物和/或第三化合物,配制成长效肠胃外施用(lap)组合物,解决了无依从性以及hiv的预防或治疗的问题,所述lap组合物适合于例如每月一次,每2个月一次,每3个月一次,每6个月一次或每12个月一次的施用。例如,本发明的lap组合物可以包含药学上可接受的赋形剂和具有以下结构的第一化合物:(“第一化合物”)或其药学上可接受的盐,结合:具有以下结构的第二化合物(“第二化合物”):或其药学上可接受的盐,以及任选地,还结合具有以下结构的第三化合物(“第三化合物”):。在其它实施方案中,所述第一化合物和第二化合物可包含其异构体形式,其中本发明提供长效肠胃外(lap)药物组合物,其包含药学上可接受的赋形剂和具有以下结构的第一化合物:或其药学上可接受的盐,结合具有以下结构的第二化合物:或其药学上可接受的盐。在本发明的一个方面,提供了包含第一化合物、第二化合物、和/或第三化合物、或它们的药学上可接受的盐的lap药物组合物。在本发明的第二方面,提供了用于在患有hiv感染的人中治疗或预防hiv感染的方法,包括向该人施用lap药物组合物,所述lap药物组合物包括第一化合物、第二化合物、和/或第三化合物、或它们的药学上可接受的盐。在本发明的第三方面,提供了用于在患有hiv感染的人中治疗或预防hiv感染的方法,包括向该人施用lap药物组合物,所述lap药物组合物包括第一化合物、第二化合物、和/或第三化合物、或它们的药学上可接受的盐。在本发明的第四方面,提供了包含第一化合物、第二化合物、和/或第三化合物、或它们的药学上可接受的盐的lap药物组合物在hiv药物治疗中的用途。在本发明的第五方面,提供了所述第一化合物、第二化合物、和/或第三化合物、或它们的药学上可接受的盐在制备用于在人类中治疗或预防hiv感染的长效肠胃外药物中的用途。在本发明的第六方面,提供了所述第一化合物、第二化合物、和/或第三化合物、或它们的药学上可接受的盐在制备用于在人类中治疗或预防hiv感染的长效肠胃外药物中的用途。附图简述图1描绘了在5mg/kg和20mg/kg剂量的lap大鼠pk研究的第一化合物的lap平均浓度对时间(小时)的曲线。图2描绘了在2.5mg/kg和5mg/kg剂量的lap狗pk研究的第一化合物的lap平均浓度对时间(小时)的曲线。图3描绘了不同药物微粒制剂的lap大鼠(im)pk研究的第一化合物的lap平均浓度对时间(小时)的曲线。发明详述由四个蛋白结构域(基质(ma)、衣壳(ca)、核衣壳(nc)和p6)以及两个间隔肽(sp1和sp2)组成的hivgag多聚蛋白前体(pr55gag)代表了新的治疗靶标。虽然gag多聚蛋白的裂解在感染性病毒颗粒产生的进程中起核心作用,但是迄今为止还没有抗逆转录病毒药物被批准用于该机制。在大多数细胞类型中,组装发生在质膜处,并且gag的ma结构域介导了膜的结合。通过未成熟颗粒从细胞出芽来完成组装。伴随着颗粒释放,病毒编码的pr将gag裂解成四个成熟蛋白结构域,ma、ca、nc和p6,以及两个间隔肽sp1和sp2。pr还裂解了gag-pol,释放出病毒酶pr、rt和in。gag蛋白酶解加工诱导颗粒内的形态重排,称为成熟。成熟将未成熟的环形颗粒转化为成熟毒粒,该成熟毒粒包含由ca壳组成的致密锥形核,所述ca壳包围与nc络合的病毒rna基因组以及病毒酶rt和in。成熟制备病毒以感染新的细胞,并且对于粒子感染力是绝对必要的。贝韦立马(bevirimat,pa-457)是一种成熟抑制剂,其抑制gag加工中的最后一步,形成感染性病毒颗粒所需的衣壳-sp1(p25)向衣壳的转化。贝韦立马对耐art和野生型hiv具有活性,并已显示与所有类别的抗逆转录病毒药物的协同作用。在达到≥20μg/ml的谷底水平并且在q369、v370或t371处没有任何关键性基线gag多态性的患者中,贝韦立马将hiv病毒载量平均降低1.3log10/ml。然而,在q369、v370或t371处具有gag多态性的贝韦立马使用者显示出的载量降低与在这些位点没有gag多态性的患者相比显著更低。可以在下列专利申请中找到成熟抑制剂的其他实例:pct专利申请wo2011/100308,“derivativesofbetulin”;pct专利申请pct/us2012/024288,“novelanti-hivcompoundsandmethodsofusethereof”;中国pct申请pct/cn2011/001302,“carbonylderivativesofbetulin”;中国pct申请pct/cn2011/001303,“methylenederivativesofbetulin”;中国pct申请pct/cn2011/002105和pct/cn2011/002159,“propenoatederivativesofbetulin”;和美国临时申请61/576,448,“derivativesofbetulin”。随着成熟抑制剂的每次迭代,需要优化多态性分离物优势度并实现最大效力,同时使蛋白质移位(proteinshift)最小化。到目前为止,还没有成熟抑制剂实现了这三种性质的最佳平衡。源自2011年12月16日提交的美国临时申请61/576448的已公开的pct申请wo2013090664(其通过引用整体并入本文)公开了成熟抑制剂,其是可用于治疗hiv感染和艾滋病的桦木醇衍生物。这些桦木醇衍生物包括为“第一化合物”的4-(((3ar,5ar,5br,7ar,9s,11ar,11br,13as)-3a-((r)-2-((4-氯苄基)(2-(二甲基氨基)乙基)氨基)-1-羟基乙基)-1-异丙基-5a,5b,8,8,11a-五甲基-2-氧代-3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a-十八氢-2h-环戊二烯并[a]䓛-9-基)氧基)-2,2-二甲基-4-氧代丁酸:是被认为提供多态性分离物优势度的最优化的成熟抑制剂,其实现最大效力,同时使蛋白质移位最小化。该化合物目前正被开发用于治疗hiv感染和相关疾病状态。“第二化合物”是cabotegravir,一种hiv整合酶抑制剂,目前在由glaxosmithkline开发作为长效肠胃外药物。第二化合物具有以下结构:。技术人员将从以下已公开的pct申请理解如何制备cabotegravir:pct专利申请pct/us2006/016604和pct专利申请pct/us2011/051713,它们两者都通过引用整体并入本文。“第三化合物”是利匹韦林(rilpivirine),4-[[4-[[4-(2-氰基乙烯基)-2,6-二甲基苯基]氨基]-2-嘧啶基]氨基]苄腈,其是人免疫缺陷病毒1型(hiv-1)的非核苷类逆转录酶抑制剂(nnrti),并且被表明与其它抗逆转录病毒药物组合用于治疗初次接受治疗的成年患者中的hiv-1感染。盐酸利匹韦林作为薄膜包衣片剂在欧洲和美国(商品名edurant)被推出。本领域技术人员将从以下美国专利6838464、7067522、7125879、7638522、8080551和8101629中的一个或多个理解如何制备利匹韦林,所有这些文献通过引用整体并入本文。利匹韦林(即第三化合物)具有以下结构:。尽管在过去十年中已经取得了重大进展来抑制hiv-1的复制,从而防止aids的临床表现,但是目前可用的hiv感染治疗中没有一种可以治愈感染。此外,haart或由至少三种抗逆转录病毒药物组成的高活性抗逆转录病毒治疗可能在病毒抗性发展后失败。导致hiv不完全抑制和抗性发展的因素包括药物效力不足、不依从性、组织穿透受限、耐药性和几种宿主因素,如宿主遗传因素。因此,在终身治疗期间的依从性是至关重要的,因为在血液中建立最小抑制药物浓度抑制病毒生长和抗性株的发展。本发明通过将第一化合物、第二化合物和任选的第三化合物各自分别或它们的两个或三个一起配制为长效肠胃外(lap)组合物或贮库制剂来解决治疗或预防hiv中的这些问题,所述长效肠胃外(lap)组合物或贮库制剂适合于,例如,每周施用一次,每两周施用一次,每月施用一次,每2个月施用一次,每3个月施用一次,每6个月施用一次或每12个月施用一次。所述第一化合物、第二化合物和任选的第三化合物的长效肠胃外制剂可以以不频繁的给药产生持续有效的抑制浓度,并且可以改善对治疗的依从性。除了在传统抗-hiv治疗之后有利于维持病毒抑制之外,长效制剂也可以用作暴露前预防的实践机会。本发明的特征在于适于每月或更长时间施用一次的包含活性成分的药物组合物,所述活性成分是第一和第二化合物和任选的第三化合物、或它们的药学上可接受的盐。本发明的其它特征是使用这些药物组合物的方法。在一个实施方案中,本发明的特征在于药物组合物,所述药物组合物包含第一化合物、第二化合物、和/或第三化合物、或它们的药学上可接受的盐、以及表面活性剂体系。在其它实施方案中,本发明的特征在于包含治疗有效量的第一化合物和第二化合物和任选的第三化合物、或它们的药学上可接受的盐、以及表面活性剂体系的药物组合物。药学上可接受的盐包括,但不限于,描述于源自2011年12月16日提交的美国临时申请61/576448的已公开的pct申请wo2013090664中的那些。本文所用的术语“治疗有效量”是指减少或逆转或治疗人或其它哺乳动物中的疾病的足够量的药物、化合物、组合物、产品或药剂。本发明的特征在于用于向受试者(例如人)施用的肠胃外药物组合物。在另一个实施方案中,本发明的特征在于用于每周(每周一次)施用的包含第一化合物和第二化合物和任选的第三化合物、或它们的药学上可接受的盐、以及表面活性剂体系的长效肠胃外药物组合物。在另一个实施方案中,本发明的特征在于用于每两周(每两周一次)施用的包含第一化合物和第二化合物和任选的第三化合物、或它们的药学上可接受的盐、以及表面活性剂体系的长效肠胃外药物组合物。在另一个实施方案中,本发明的特征在于用于每月(每月一次)施用的包含第一化合物和第二化合物和任选的第三化合物、或它们的药学上可接受的盐、以及表面活性剂体系的长效肠胃外药物组合物。在另一个实施方案中,本发明的特征在于用于每两月(每两月一次)施用的包含第一化合物和第二化合物和任选的第三化合物、或它们的药学上可接受的盐、以及表面活性剂体系的长效肠胃外药物组合物。在另一个实施方案中,本发明的特征在于用于每三月(每三月一次)施用的包含第一化合物和第二化合物和任选的第三化合物、或它们的药学上可接受的盐、以及表面活性剂体系的长效肠胃外药物组合物。在另一个实施方案中,本发明的特征在于用于每六个或十二个月或在该范围内的任何时间点施用一次的包含第一化合物和第二化合物和任选的第三化合物、或它们的药学上可接受的盐、以及表面活性剂体系的长效肠胃外药物组合物。本发明的组合物提供第一和第二化合物和任选的第三化合物在受试者体内延长的时间段内的缓慢释放。因此,为了达到药物的治疗水平,第一和第二化合物和任选的第三化合物有利地在约一至三个月或在该范围内的任何时间点内从组合物中释放。本发明的一个实施方案是适于肠胃外施用的药物组合物,其包含第一和第二化合物和任选的第三化合物,以及为第一和第二化合物和任选的第三化合物提供在一周至三个月的时期内释放的包含聚合物的组合的表面活性剂体系。聚合物的一个合适组合是,例如,聚山梨醇酯80和聚乙烯吡咯烷酮(pvp)。本发明的组合物可以通过多种途径,包括肌内(im)、静脉内(iv)或皮下(sq),施用于受试者。因此,在一个实施方案中,本发明的组合物通过肌内途径施用于受试者。在另一个实施方案中,本发明的组合物通过静脉内途径施用于受试者。在另一个实施方案中,本发明的组合物通过皮下途径施用于受试者。为了实现本发明的目的,“表面活性剂体系”是指包括至少一种表面活性剂的任何适于药物目的的配方。例如,除了表面活性剂之外,可以与本发明一起使用的表面活性剂体系可以包括另外的组分,例如缓冲剂、聚合物(用于药物颗粒)、润湿剂、稳定剂、张力调节剂和溶剂(如水)。表面活性剂体系可以包括任何表面活性剂,只要其与药物应用相容即可。例如,合适的表面活性剂包括,但不限于,聚氧乙烯脱水山梨糖醇脂肪酸酯(聚山梨醇酯,例如聚山梨醇酯20或80),泊洛沙姆(例如作为环氧乙烷和环氧丙烷的嵌段共聚物的lutrol™f68、f108和f127,十二烷基硫酸钠和/或月桂基硫酸钠),脂肪酸的脱水山梨糖醇酯(span),聚乙氧基化蓖麻油及其衍生物,生育酚聚乙二醇琥珀酸酯和聚乙烯醇。在某些实施方案中,所述表面活性剂体系包含的表面活性剂的量在约0.01%(w/v)至约5%(w/v)表面活性剂的范围。在其它实施方案中,所述表面活性剂体系包含的表面活性剂的量在约0.1%(w/v)至约3%(w/v)表面活性剂的范围。在另外的其它实施方案中,所述表面活性剂体系包含约0.2%(w/v)的表面活性剂。在另外的其它实施方案中,所述表面活性剂体系包含约0.4%(w/v)的表面活性剂。在其它实施方案中,所述表面活性剂体系包含聚山梨醇酯-80(例如,吐温-80)。在另外的其它实施方案中,所述表面活性剂体系包含0.4%(w/v)的聚山梨醇酯-80。代表性的稳定剂包括,但不限于,聚乙二醇,羧甲基纤维素钙,羧甲基纤维素钠,甲基纤维素,羟乙基纤维素,羟丙基纤维素,羟甲基丙基纤维素,多糖,透明质酸,聚乙烯醇(pva)和聚乙烯吡咯烷酮(pvp)。在某些实施方案中,所述表面活性剂体系包含的稳定剂的量在约0.01%(w/v)至约5%(w/v)稳定剂的范围。在其它实施方案中,所述表面活性剂体系包含的稳定剂的量在约1%(w/v)至约5%(w/v)稳定剂的范围。在其它实施方案中,所述表面活性剂体系包含的稳定剂的量在约1%(w/v)至约3%(w/v)稳定剂的范围。在另外的其它实施方案中,所述表面活性剂体系包含约2%(w/v)的稳定剂。在其它实施方案中,所述表面活性剂体系包含聚乙二醇。在其它实施方案中,所述表面活性剂体系包含peg-3350。在另外的其它实施方案中,所述表面活性剂体系包含2%(w/v)的peg-3350。合适的缓冲盐包括,但不限于,选自磷酸盐、柠檬酸盐、乙酸盐和酒石酸盐等的缓冲盐。在某些实施方案中,所述表面活性剂体系包含的缓冲盐的量在约1mm至约100mm缓冲盐的范围。在其它实施方案中,所述表面活性剂体系包含的缓冲盐的量在约2mm至约50mm缓冲盐的范围。在其它实施方案中,所述表面活性剂体系包含的缓冲盐的量在约3mm至约25mm缓冲盐的范围。在其它实施方案中,所述表面活性剂体系包含的缓冲盐的量在约5mm至约15mm缓冲盐的范围。在另外的其它实施方案中,所述表面活性剂体系包含约10mm缓冲盐。在某些实施方案中,将缓冲盐的ph调节至约ph6.0至约ph8.0的范围。在其它实施方案中,将缓冲盐的ph调节至约ph6.5至约ph7.5的范围。在其它实施方案中,将缓冲盐的ph调节至约ph6.7至约ph7.3的范围。在一个实施方案中,所述缓冲盐包括磷酸盐缓冲盐水(pbs)。在另一个实施方案中,所述缓冲盐包含浓度为约10mm的磷酸盐缓冲盐水。在另一个实施方案中,所述缓冲盐包含浓度为约10mm和ph为约6.9的磷酸盐缓冲盐水。合适的张力调节剂包括,但不限于,氯化钠,甘露醇,蔗糖,麦芽糖和右旋糖等。在一个实施方案中,张力调节剂包括氯化钠。在另一个实施方案中,张力调节剂为氯化钠。在某些实施方案中,所述表面活性剂体系包含的张力调节剂的浓度在约0至约350mm的范围。在某些实施方案中,所述表面活性剂体系包含的张力调节剂的浓度在约0至约175mm的范围。在某些实施方案中,所述表面活性剂体系的张力在约250至约350mosmol/kg的范围。在一个实施方案中,第一和第二化合物和任选的第三化合物可以作为微粒悬浮在表面活性剂体系和水性缓冲液中。在一些实施方案中,第一化合物可以是无定形形式或结晶形式。通常,药物粒度(d50)将在约0.05μm至约100μm的范围内。在其它实施方案中,药物粒度将在约0.1μm至约50μm的范围内。在其它实施方案中,药物粒度将在约0.1μm至约20μm的范围内。在其它实施方案中,药物粒度(d50)将在约0.1μm至约10μm的范围内。在其它实施方案中,药物粒度(d50)将在约0.1μm至约5μm的范围内。在其它实施方案中,药物粒度(d50)将在约1μm至约5μm的范围内。在其它实施方案中,药物粒度(d50)将在约0.05μm至约0.05μm的范围内。在其它实施方案中,药物粒度(d50)将在约0.5μm至约5μm的范围内。在其它实施方案中,药物粒度(d50)将在约5μm至约25μm的范围内。在其它实施方案中,药物粒度(d50)将在约25μm至约100μm的范围内。在另外的其它实施方案中,表面活性剂体系中的药物粒度可以是混合的尺寸。例如,具有从相对较大到相对较小的基本上不同的粒度,可以实现制剂的可接受的药代动力学参数,因为小颗粒比大颗粒更快地被吸收和代谢。这种类型的混合粒度制剂可以通过在施用后早期向受试者提供药物的快速释放,同时在施用后在长时间内仍然保持药物的长效释放,来增强本发明的长效性质。因此,在一个实施方案中,本发明的lap发明可以包括两种或更多种基本上不同的粒径,其允许第一化合物和第二化合物以及任选的第三化合物的更早和更晚的释放,并且这种不同的吸收动力学将是增强持久长效的药物暴露的手段。在一个实施方案中,第一化合物为微粒形式,其中第一化合物的微粒尺寸范围为约0.05μm至约100μm,其中所述微粒包含两种或更多种基本上不同的粒径。在另外的其它实施方案中,第一和第二化合物和任选的第三化合物的药物颗粒被包封到基于聚合物的微粒中,其可以任选地随后冷冻干燥用于长期储存。当关于本发明使用术语“包封”时,是指第一和第二化合物和任选的第三化合物基本上被聚合物包围,尽管一些化合物仍然可以存在于所述包封化合物/聚合物结构的表面。在使用之前,可以任选立刻将干燥的微粒悬浮在缓冲水溶液中。用于制备这种微粒的聚合物可以选自一系列可生物降解的聚合物,包括聚(乳酸-共-乙醇酸)(mw5-200kd)及其衍生物,例如基于聚乙二醇的两亲性聚合物等。微粒尺寸(d50)可以在约1μm至约100μm的范围内,并且药物包封可以在约10%至约70%(w/w)的范围内。在一个实施方案中,将第一和第二化合物和任选的第三化合物的药物颗粒包封到基于聚合物的微粒(例如含有resomer™的那些微粒)中。在另一个实施方案中,将第一和第二化合物和任选的第三化合物的药物颗粒包封到基于聚合物的微粒(例如含有resomer™752s的那些微粒)中。在其它实施方案中,原位凝胶可用于包封第一化合物。这可以是包含第一化合物和水不溶性的凝胶形成聚合物的基于水混溶性有机溶剂的溶液。一旦施用(im或sc),有机溶剂就会消散,而水不溶性聚合物会沉淀出来以形成含有第一化合物的凝胶。然后随着基于该聚合物的凝胶在体内降解,第一化合物将缓慢扩散出来。用于制备原位凝胶的聚合物选自一系列可生物降解的聚合物,包括聚(乳酸-共-乙醇酸)(mw5-200kd)及其衍生物,基于聚乙二醇的两亲性聚合物等。所述有机溶剂选自n-甲基吡咯烷酮(nmp)、二甲亚砜(dmso)、二甲基甲酰胺(dmf)、二甲基乙酰胺(dma)等。所述聚合物在所述有机溶剂中的浓度可以在1-50%(w/w)之间,而第一化合物的浓度可以在1-50%(w/w)之间。或者,所述微粒制剂可以通过喷雾干燥方法制备。类似地,将如本文所述制备的含有第一化合物和所选择的聚合物的有机溶液进行喷雾干燥过程,其中有机溶剂在氮气流下快速蒸发以形成包封第一和第二化合物和任选的第三化合物的微粒。干燥温度不低于35℃,并且溶液喷雾速率不小于0.1ml/min。对于原位凝胶微粒,第一化合物和选择的聚合物可以共溶解在合适的有机溶剂中,其中有机溶剂必须满足以下标准:a)对所选择的聚合物具有良好的溶解性;b)与水溶液具有良好的混溶性;和c)当在人中使用时具有低毒性和已证明的安全性;例如n-甲基吡咯烷酮(nmp)、二甲亚砜(dmso)、二甲基甲酰胺(dmf)、二甲基乙酰胺(dma)等。可通过改变在溶剂中的聚合物浓度、聚合物对第一化合物的比率来配制所得的含第一化合物和所选聚合物的溶液,以便控制施用后的凝胶形成速率和随后的药物扩散速率。该溶液最终通过以25kgy的最小剂量在干冰上进行γ照射来进行最终灭菌。聚合物的组合的实例包括作为润湿剂的聚山梨醇酯(例如聚山梨醇酯80)和作为稳定剂的聚乙烯吡咯烷酮(pvp)(例如plasdonek29/32)。因此,在一个实施方案中,本发明的特征在于一种肠胃外药物组合物,其包含第一和第二化合物和任选的第三化合物、或它们的药学上可接受的盐,和聚山梨酯80和聚乙烯吡咯烷酮:plasdonek29/32。本发明的一个实施方案是一种用于肠胃外施用的药物组合物,其包含第一和第二化合物和任选的第三化合物,以及适用于通常已知的灭菌技术如γ照射、电子束照射和高压灭菌的表面活性剂体系。本发明的一个实施方案是一种用于肠胃外施用的药物组合物,其包含第一和第二化合物和任选的第三化合物,以及可使用无菌技术制备的表面活性剂体系。本发明的一个实施方案是一种用于肠胃外施用的药物组合物,其包含第一和第二化合物和任选的第三化合物,以及适合于γ辐射灭菌的表面活性剂体系。本发明的一个实施方案是一种用于肠胃外施用的药物组合物,其包含第一和第二化合物和任选的第三化合物,以及适合于通过电子束照射或高压灭菌消毒的灭菌技术的表面活性剂体系。本发明的一个实施方案是一种用于肠胃外施用的药物组合物,其可以以“即用型”无菌悬浮液或冻干剂的形式用于重新构成。本发明的组合物可以通过皮下或肌肉内注射施用。本发明的组合物还可以通过皮内或玻璃体内注射或植入来施用。本发明的组合物还可以通过其它肠胃外施用途径施用。本发明的组合物的制备可以通过使用湿珠磨机研磨并通过γ照射灭菌来进行。本发明的另一个特征是简化hiv的治疗方案,目的是通过提供简化剂型来增强患者依从性,所述简化剂型含有治疗有效量的第一和第二化合物和任选的第三化合物、或它们药学上可接受的盐。本发明的特征还在于用于治疗人类中的hiv感染的方法,所述方法包括向所述人施用根据本发明的组合物。本发明的特征在于根据本发明的药物组合物在治疗hiv感染中的用途。本发明的特征在于制备用于医学治疗的根据本发明的药物。本发明的特征在于制备用于治疗hiv感染的根据本发明的药物。本发明的特征还在于治疗人类hiv感染的方法,所述方法包括在以片剂或溶液形式用第一和第二化合物和任选的第三化合物治疗之前、期间或之后给所述人施用根据本发明的组合物。本领域技术人员将理解,本文提及的“治疗”延伸到治疗已确立的疾病、感染或其症状。本发明的特征还在于用于预防人类中的hiv感染的方法,所述方法包括向所述人施用根据本发明的组合物。本发明的特征在于根据本发明的药物组合物在预防hiv感染中的用途。本发明的特征在于制备用于预防性医学治疗的根据本发明的药物。本发明的特征在于制备用于预防hiv感染的根据本发明的药物。本发明的特征还在于治疗或预防人类hiv感染的方法,所述方法包括在以片剂或溶液形式用第一和第二化合物和任选的第三化合物治疗之前、期间或之后给所述人施用根据本发明的组合物。因此,在本发明的某些实施方案中,提供了一种药物组合物,其包含治疗有效量的长效制剂,所述长效制剂包含具有以下结构的第一化合物:(“第一化合物”)或其药学上可接受的盐,结合:具有以下结构的第二化合物(“第二化合物”):或其药学上可接受的盐,以及任选地结合具有以下结构的第三化合物(“第三化合物”):在用于肠胃外施用的药学上可接受的载体中。在其它实施方案中,提供了包含第一和第二化合物和任选的第三化合物的药物组合物,其被配制用于皮下施用。在其它实施方案中,提供了包含第一和第二化合物和任选的第三化合物的药物组合物,其被配制用于肌肉内施用。在其它实施方案中,提供了包含第一和第二化合物和任选的第三化合物的药物组合物,其被配制为每周或更长时间施用一次。在其它实施方案中,提供了包含第一和第二化合物和任选的第三化合物的药物组合物,其被配制为每周施用一次。在其它实施方案中,提供了包含第一和第二化合物和任选的第三化合物的药物组合物,其被配制为每月施用一次。在其它实施方案中,提供了包含第一和第二化合物和任选的第三化合物的药物组合物,其被配制为每两月施用一次。在其它实施方案中,提供了包含第一和第二化合物和任选的第三化合物的药物组合物,其被配制为每三月施用一次。在其它实施方案中,提供了包含第一和第二化合物和任选的第三化合物的药物组合物,其被配制为在30天和365天之间的任何间隔施用一次。在其它实施方案中,提供了包含第一化合物和第二化合物和任选的第三化合物的药物组合物,其中所述化合物以结晶纳米颗粒的形式存在于组合物中。在其它实施方案中,提供了包含第一化合物和第二化合物和任选的第三化合物的药物组合物,其中所述化合物以基质释放颗粒的形式存在于组合物中。在其它实施方案中,提供了包含第一化合物和第二化合物和任选的第三化合物的药物组合物,其中所述组合物可以通过γ照射最终灭菌。在其它实施方案中,提供了用于治疗患有hiv感染的人中的hiv感染的方法,包括向该人施用单一治疗药物组合物,其包含治疗有效量的长效制剂,该长效制剂包含第一和第二化合物和任选的第三化合物、或它们的药学上可接受的盐,在用于肠胃外施用的药学上可接受的载体中。在其它实施方案中,提供了用于预防人中的hiv感染的方法,包括向有风险获得hiv感染的人施用单一治疗药物组合物,其包含治疗有效量的长效制剂,该长效制剂包含第一化合物和第二化合物和任选的第三化合物或它们药学上可接受的盐,在用于肠胃外施用的药学上可接受的载体中。在其它实施方案中,提供了一种lap药物组合物,其包含第一化合物、第二化合物、和/或第三化合物、或它们的药学上可接受的盐。在其它实施方案中,提供了用于在患有hiv感染的人中治疗hiv感染的方法,包括向该人施用lap药物组合物,所述lap药物组合物包括第一化合物、第二化合物、和/或第三化合物、或它们的药学上可接受的盐。在其它实施方案中,提供了用于在患有hiv感染的人中预防hiv感染的方法,包括向该人施用lap药物组合物,所述lap药物组合物包括第一化合物、第二化合物、和/或第三化合物、或它们药学上可接受的盐。在其它实施方案中,提供了一种lap药物组合物,其包含:第一化合物、第二化合物和/或第三化合物、或它们的药学上可接受的盐,并且还包含表面活性剂体系。在其它实施方案中,提供了一种lap药物组合物,其包含:第一化合物和第二化合物和/或任选的第三化合物或它们的药学上可接受的盐,其还包含表面活性剂体系,其中所述表面活性剂体系包含的表面活性剂的量在约0.1%(w/v)至约3%(w/v)表面活性剂的范围,或在0.2%(w/v)至约0.4%(w/v)表面活性剂的范围,或者所述表面活性剂体系包含约0.4%(w/v)表面活性剂。在其它实施方案中,提供了一种lap药物组合物,其包含:第一化合物和第二化合物和/或任选的第三化合物,以及选自度鲁特韦(dolutegravir)和利托那韦(ritonavir)的一种或多种另外的化合物或它们药学上可接受的盐。在其它实施方案中,提供了用于在患有hiv感染的人中治疗hiv感染的方法,包括向该人施用一种lap药物组合物,该lap药物组合物包含:第一化合物和第二化合物和/或任选的第三化合物、或它们的药学上可接受的盐,以及选自度鲁特韦(dolutegravir)和利托那韦(ritonavir)的一种或多种另外的化合物或它们药学上可接受的盐。在其它实施方案中,提供了一种lap药物组合物,其包含:第一化合物和第二化合物和/或第三化合物、或它们药学上可接受的盐,以及任何增强剂(例如利托那韦)。所述增强剂可以在相同iv或sc注射器中与第一化合物同时施用,或者它可以以口服片剂或胶囊单独施用。在其它实施方案中,包含第一化合物和第二化合物和/或任选的第三化合物的lap组合物仅在受试者已经施用包含普遍接受的抗逆转录病毒(arv)方案的治疗后施用给受试者。初始arv方案通常由与nnrti、pi(优选用利托那韦[rtv]增强)、insti、或ccr5拮抗剂(即马拉韦罗(maraviroc)[mvc])组合的两种nrti组成。在临床试验中,基于nnrti、pi、insti或ccr5拮抗剂的方案在大多数患者中都导致hivrna减少和cd4细胞增加。例如,对于初次接受抗逆转录病毒(arv)治疗的患者,一种普遍接受的arv方案可以选自以下任何一种:依法韦仑(efavirenz)/富马酸替诺福韦酯(tenofovirdisoproxilfumarate)/恩曲他滨(emtricitabine)(efv/tdf/ftc)利托那韦增强的阿扎那韦(atazanavir)+富马酸替诺福韦酯/恩曲他滨(atv/r+tdf/ftc)利托那韦增强的达芦那韦(darunavir)+富马酸替诺福韦酯/恩曲他滨(drv/r+tdf/ftc)拉替拉韦(raltegravir)+富马酸替诺福韦酯/恩曲他滨(ral+tdf/ftc)。本发明的药物组合物以适于肠胃外施用的药物组合物形式提供。所述组合物还可以包括安全且有效量的其它活性成分,例如抗微生物剂、抗病毒剂或防腐剂。本领域技术人员应当理解,用于治疗所需的活性成分的量将根据多种因素而变化,包括所治疗的病症的性质和患者的年龄和状况,并且最终由主治医师,兽医或保健医生决定。本发明的组合物使得患者能够免于多剂量方案而得到更多自由,并且减轻了记住复杂的每日施用时间和计划表所需的辛苦。本发明的组合物特别适合于作为单剂量每月、每两个月或每三个月或以30至365天之间的任何间隔(包括每六个月或十二个月)施用。有利地,本发明的组合物可以每月施用一次。本发明的组合物作为多药物治疗方案的组分可以与其它药物制剂组合使用。这样的组合可以以一个剂量单位(例如固定剂量组合)施用于受试者,或者其可以以单独的剂量单位施用。本发明的组合物还可以包装为制品,所述制品包含:治疗有效量的式(i)化合物或其药学上可接受的盐;和治疗有效量的一种或多种以下药物:核苷逆转录酶抑制剂,非核苷逆转录酶抑制剂,蛋白酶抑制剂和整合酶抑制剂。包装材料还可以具有在其上印刷的与所述药物组合物相关的标签和信息。此外,制品可以包含含有产品信息的说明书、报告、通知、小册子或传单。这种形式的药物信息在制药工业中被称为“包装插页”。包装插页可以附着于或包括在药品制品中。包装插页和任何制品标签提供了与药物组合物有关的信息。所述信息和标签提供了医疗保健专业人员和患者使用的各种形式的信息,描述了管理机构(如美国食品和药物管理局)要求的组成、其剂量和各种其它参数。本发明还提供了以下实施方案:(a)肠胃外药物组合物,其包含有效量的第一和第二化合物和/或任选的第三化合物、或它们药学上可接受的盐,用于长期治疗hiv感染或在有被hiv感染风险的个体中防止hiv感染,其中所述组合物以至少一周的时间间隔间歇地施用。(b)根据(a)的组合物,其中所述组合物每两周施用一次。(c)根据(a)的组合物,其中所述组合物每月施用一次。(d)根据(a)至(c)中任一项的组合物,其中,选择第一和第二化合物和/或任选的第三化合物、或它们药学上可接受的盐中的每一种的有效量,使得在受试者中的第一和第二化合物和/或任选的第三化合物的血浆浓度在延长的时间段内保持在最大血浆水平(其是引起显著副作用的血浆水平)和最小血浆水平(其是使得第一和第二化合物和任选的第三化合物提供有效治疗或预防hiv感染的最低血浆水平)之间的水平。(e)根据(d)的组合物,其中,将受试者的血浆水平保持在等于或高于约150ng/ml,特别是等于或高于约600ng/ml的水平。(f)根据(a)至(e)中任一项所述的组合物,其中,所述组合物通过皮下或肌肉内施用。(g)根据(a)至(f)中任一项所述的组合物,其包含上述包含聚山梨醇酯和/或聚乙烯吡咯烷酮的表面活性剂体系。(h)一种治疗或预防人hiv感染的方法,包括根据上述(a)至(g)中任一项的药物组合物。可以选择施用的第一和第二化合物和任选的第三化合物的剂量(其是用于本发明的肠胃外组合物中的化合物的量),以使得所述化合物在受试者中的血浆浓度在延迟的时间段内保持高于最小血浆水平。在本文中,术语“最小血浆水平”(或cmin)是指最低的有效血浆水平,即提供有效预防或治疗hiv感染的化合物血浆水平。在hiv从感染hiv的个体传播到未感染hiv的个体的情况下,这是有效抑制所述传播的最低血浆水平。受试者中的包含第一和第二化合物和任选的第三化合物的lap制剂的血浆水平可以保持在约170ng/ml、约700ng/ml或约1000ng/ml的高于最低血浆水平的水平。受试者中的所述化合物的血浆水平可以保持在这些最低血浆水平以上,因为在更低水平时,药物可能不再有效,从而增加hiv感染的传播风险,并且可能不适合治疗hiv感染对象。所述化合物的血浆水平可以保持在更高水平以避免hiv突变的发展,同时保持安全界限。包含第一和第二化合物和(i)的lap制剂的施用模式的优点是可以实现高cmin水平而没有相应的高cmax,这可以减轻与cmax相关的潜在副作用。可以选择待施用的第一和第二化合物和任选的第三化合物的有效量,以使得受试者中的血浆浓度在延长的时间段内保持在最大血浆水平(或cmax)和最小血浆水平(或cmin)之间的水平。在一些实施方案中,受试者中所述化合物的血浆水平可以保持在最小血浆水平(或如上所述的cmin)和化合物(i)的较低的最大血浆水平(或cmax)(其被定义为对应于所述化合物在治疗上起作用的最低血浆水平的水平)之间。所述化合物在治疗上起作用的最低水平是有效抑制hiv在感染hiv的个体中的复制的最低血浆水平,以使得hiv的病毒载量相对较低,例如其中病毒载量(表示为病毒rna在指定体积的血清中的拷贝的数量)低于约200拷贝/ml,特别是低于约100拷贝/ml,更特别是低于50拷贝/ml,特别是低于hiv检测的检测限。如上所述,所述化合物的血浆水平取决于施用的每次肠胃外剂量中活性成分的量。然而,它也取决于使施用的频率(即每次施用之间的时间间隔)。两个参数可用于将血浆水平引导至期望值。当施用不太频繁时,剂量可以更高。尽管所述化合物的血浆水平应保持低于最大值或高于最小值,但它们可能在相对短的时间段(其可以为尽可能短)内超过最大值或降至最小值以下。因此,最大和最小血浆水平可以表示为在某一时间段内的平均血浆水平。在一些情况下,在施用后不久可能有小的初始血浆浓度峰值,之后血浆水平达到稳态。本发明的组合物方便地允许以单位剂型施用第一和第二化合物和任选的第三化合物,所述单位剂型含有,例如,约1mg至约1000mg,约20mg至约100mg,约20mg至约300mg,约25mg至约800mg,约25mg至约100mg,约100mg至约200mg,约200mg至约400mg,约100mg至约800mg,约100mg至约600mg,约100mg至约400mg/单位剂型,或约400mg至约800mg。在一个实施方案中,单位剂量为约100mg至约200mg,其每月一次施用于受试者。在一些实施方案中,可以存在大体上高于较后维持剂量的初始负荷剂量。因此,在一个实施方案中,第一化合物最初以范围为400mg至800mg的量的负载剂量施用于受试者,然后以范围为约20mg至约300mg的量的作为维持剂量施用。在另一个实施方案中,受试者可以最初以800mg给药,然后以100mg给药。在所述制剂中的第一和第二化合物和任选的第三化合物的单位剂量浓度可以选自以下范围中的任一个:0.05-0.5μm,0.5-1μm,1-5μm,5-25μm,25-50μm,或50-150μm。可以基于约1mg/天至约50mg/天,优选3mg/天至约30mg/天计算待施用的剂量。这对应于约7mg至约350mg,优选约20mg至约200mg的每周剂量,或对应于约30mg至约1500mg,优选约90mg至约900mg的每月剂量。其它给药方案的剂量可以通过将每日剂量与每次给药之间的天数相乘来容易地计算。可以基于约0.001mg/kg/天至约1mg/kg/天,优选0.05mg/kg/天至约0.5mg/kg/天计算待施用的剂量。这对应于约0.5mg至约500mg,优选约20mg至约200mg的每周剂量,或对应于约30mg至约1500mg,优选约90mg至约900mg的每月剂量。其它给药方案的剂量可以通过将每日剂量与每次给药之间的天数相乘来容易地计算。一旦施用,受试者中第一和第二化合物和任选的第三化合物的血浆水平可以或多或少是稳定的。在血浆水平的初始上升之后,可以在延长的时间段内实现稳态模式。“稳态”是指其中在受试者的血浆中存在的药物的量在延长的时间段内保持在大致相同水平的状况。随后,第一和第二化合物和任选的第三化合物的血浆水平可以随时间逐渐降低,并且当达到最小血浆水平时,可以施用下一剂量的第一和第二化合物和任选的第三化合物。术语“保持在大致相同水平”不排除在可接受范围内,例如在约30%,约20%或约10%内,可存在血浆浓度的小波动。第一和第二化合物和任选的第三化合物的肠胃外组合物可以通过静脉内注射或优选通过皮下或肌内施用来施用。本发明基于使用包含第一和第二化合物和任选的第三化合物的活性成分的肠胃外组合物,因此选择载体的性质以适合肠胃外施用。在大多数情况下,载体将包括无菌水,尽管可以包括其它成分,例如来帮助溶解。例如,可以制备注射液或悬浮液,其中载体包括盐水、葡萄糖溶液或盐水和葡萄糖溶液的混合物。此外,载体可以包含上述表面活性剂体系,例如聚山梨醇酯和聚乙二醇。本发明的包含第一和第二化合物和任选的第三化合物的肠胃外药物组合物是长效的。因此,与常规组合物或与化学结构上与第一和第二化合物和任选的第三化合物相似的其它化合物相比,所述组合物可以以长的时间间隔施用用于治疗或预防hiv感染。本发明的组合物可间歇地施用于患者,例如每周一次,每月一次,每2个月一次或每3个月一次。在一个实施方案中,本发明的组合物可以对于第一至三月以较高剂量(例如800mg)作为“负荷剂量”施用,而在第一月至第几个月后,可以降低剂量。因此,本发明的组合物和通过使用其的皮下(sc)或肌内(im)注射的施用可以导致药物(药丸)负担或患者依从性困难的显著减少。此外,本发明的组合物的这种间歇给药可有助于将治疗保持在适当的依从性,这导致防止耐药性hiv的出现并且维持治疗效果延长的时间段。在实施方案中,第一化合物制剂是用于肌内或皮下注射施用的液体悬浮液形式,浓度范围为10mg/ml至250mg/ml,并且注射体积最高达4ml(例如2次注射,每次2ml)。实施例以下实施例进一步描述和举例说明了本发明范围内的具体实施方案。给出这些实施例仅用于说明,并且不应被解释为限制,因为在不脱离本发明的精神和范围的情况下许多变化是可能的。第一化合物可以由本领域技术人员通过按照源自2011年12月16日提交的美国临时申请61/576448的pct公开申请no.wo2013090664(其公开了一类可用于治疗hiv感染和艾滋病的化合物)的教导来合成。使用thermoorion9110djwp微电极和metrohmn827ph计进行ph测量。使用advanced微样品渗透压仪(micro-osmometer)3320进行渗透压测量。使用retschpm400行星式磨机进行湿式珠磨。实施例1:lap载体的制备将1.0g聚山梨醇酯80加入到0.5l的容量瓶中。将约100ml的注射用水(wfi)加入该瓶中以溶解。将8.5g的plasdonek29/32加入到该瓶中,同时另外加入300ml的wfi。用搅拌棒搅拌内容物以溶解。加入磷酸盐缓冲液:0.11039gnah2po4;0.27598gnah2po4:h2o;和0.22572gna2hpo4以及4.16389g作为等渗剂的nacl。将混合物再次搅拌以溶解,然后定容至500ml。将溶液通过0.22微米的corning滤器过滤。所得lap载体是在磷酸盐缓冲液(0.004mnah2po4和0.006mna2hpo4)中的1.7%w/vplasdonek29/32和0.2%w/v聚山梨醇酯80。实施例2:均质化的悬浮液组合物(a)用于皮下注射(sq)的在lap载体中的2.5mg/ml的第一化合物均化溶液。将17.5mg的第一化合物加入到具有卷边盖的10ml透明无菌小瓶中。加入lap载体(实施例1中制备的)至7克的重量。使用手持polytronpt1200f均质器将该溶液匀化1-2分钟,同时速度从低增加到接近最大值。然后将溶液在环境室温下搅拌。所得的题述溶液的摩尔渗透压浓度为299mosm/kg,ph为6.92。该溶液用于5mg/kg的sq注射。(b)用于sc和im(肌内)注射的在lap载体中的10.0mg/ml的第一化合物均化溶液将80mg的第一化合物加入到具有卷边盖的10ml透明无菌小瓶中。加入lap载体(实施例1中制备的)至8克的重量。使用手持polytronpt1200f均质器将该溶液匀化1-2分钟,同时速度从低增加到接近最大值。然后将溶液在环境室温下搅拌。所得的题述溶液的摩尔渗透压浓度为300mosm/kg,ph为7.25。该溶液用于5mg/kg的im注射和20mg/kg的sq注射。(c)用于sc和im(肌内)注射的在lap载体中的25.0mg/ml的第一化合物均化溶液将250mg的第一化合物加入到具有卷边盖的20ml透明无菌小瓶中。加入lap载体(实施例1中制备的)至10克的重量。使用手持polytronpt1200f均质器将该溶液匀化1-2分钟,同时速度从低增加到接近最大值。然后将溶液在环境室温下搅拌。所得的题述溶液的摩尔渗透压浓度为323mosm/kg,ph为7.68。该溶液用于2.5mg/kg的im注射和2.5mg/kg的sq注射。(d)用于im注射的在lap载体中的40.0mg/ml的第一化合物均化溶液将160mg的第一化合物加入到具有卷边盖的5ml透明无菌小瓶中。加入lap载体(实施例1中制备的)至4克的重量。使用手持polytronpt1200f均质器将该溶液匀化1-2分钟,同时速度从低增加到接近最大值。然后将溶液在环境室温下搅拌。所得的题述溶液的摩尔渗透压浓度为329mosm/kg,ph为7.87。该溶液用于20mg/kg的im注射。实施例3:湿式珠磨配方(a)制备在lap载体中的第一化合物的湿式珠磨的储备悬浮液将500mg的第一化合物称重至50ml的研磨容器中。将式i的化合物加入到具有卷边盖的10ml透明无菌小瓶中。加入lap载体(实施例1中制备的)至10克的重量,由此获得100mg/ml的悬浮液。以4倍悬浮液体积加入珠粒,并用安全带密封研磨容器。使用行星式磨机pm400以250rpm开始研磨3小时,间隔15分钟。3小时后,将研磨容器在环境室温下放置在行星式磨机中过夜。使用25mmeasypressuresyringefilterholder(筛孔尺寸:149微米)过滤珠粒。收集乳状悬浮液并用搅拌棒搅拌以消泡。所得的湿式珠磨(wbm)悬浮液的摩尔渗透压浓度为303mosm/kg,ph为7.2。该溶液用于制备下面的wbm悬浮液。(b)用于im注射的在lap载体中的10.0mg/ml的第一化合物的wbm悬浮液将0.426g实施例3(a)的wbm悬浮液加入到具有卷边盖的5ml透明无菌小瓶中。加入lap载体(实施例1中制备的)至2克的重量。旋动内容物以混合。所得题述溶液的ph为6.87。该溶液用于5mg/kg的im注射。(c)用于sq注射的在lap载体中的2.5mg/ml的第一化合物的wbm悬浮液将0.266g实施例3(a)的wbm悬浮液加入到具有卷边盖的10ml透明无菌小瓶中。加入lap载体(实施例1中制备的)至5克的重量。旋动内容物以混合。所得题述溶液的ph为6.78。该溶液用于5mg/kg的sq注射。在sprague-dawley大鼠中以5和20mg/kg剂量进行sq和im注射,同时测量t1/2,cmax,tmax和auc。结果示于表1和图1中。在图1中,人蛋白质校正的ic90=4.31ng/ml;y轴为lap浓度平均值(n=3,每个im/sq途径);t1/2iv=3.4小时;以及auc0-24iv=2.96hr*μg/ml。表1a=24天;b=57天;c=17天;以及d=42天。还在比格犬中以5和20mg/kg剂量进行sq和im注射,同时测量t1/2,cmax,tmax和auc。结果显示在表2和图2中。在图2中,人蛋白质校正的ic90=4.31ng/ml;y轴为lap浓度平均值(n=3,每个im/sq途径);t1/2iv=6.9小时;以及auc0-24iv=4.15hr*μg/ml。表2a=22天;b=40天;c=17天;以及d=36天。实施例4:大鼠lap研究的实验程序;对于一个配方,如果需要>1μm的粒径(d50),则将药物(第一化合物)直接悬浮在缓冲水溶液中,或者首先通过空气碾磨研磨成更理想的粒径,然后制备悬浮液。在这种情况下,通过将药物和缓冲溶液组分称重到合适的容器中,然后加入注射用水来制备悬浮液。然后将混合物涡旋,直到形成均匀的悬浮液,没有可见的附聚物。然后加入额外的注射用水到目标体积。或者,如果期望<1μm的粒径(d50),则首先如上所述将药物悬浮在缓冲溶液中,然后进行珠磨或微流化过程,以将粒径减小至亚微米范围。然后将制备的悬浮液以25kgy的最小剂量通过γ辐射进行最终灭菌。对于第二配方,将药物(第一化合物)和选择的包封聚合物共溶于合适的有机溶剂中,其中有机溶剂满足以下标准:a)对第一化合物和选择的聚合物具有良好的溶解性;b)不与水混溶;c)具有低沸点,因此具有良好的挥发性。合适的有机溶剂是,例如,二氯甲烷(用于本实施例),氯仿,乙酸乙酯,甲酸乙酯等。然后将溶液以1:2至1:100的体积比与含有0.1-10%(w/v)表面活性剂的水混合以形成均匀的乳液,所述表面活性剂选自聚乙烯醇(pva-本实施例中使用1%pva)、聚乙烯吡咯烷酮(pvp)、泊洛沙姆、聚山梨醇酯、聚乙氧基化蓖麻油、生育酚聚乙二醇琥珀酸酯等。然后将乳液进行真空蒸发以完全除去挥发性有机溶剂,例如在转子蒸发器(rotorvap)中。然后将均匀的悬浮液离心,并将所得颗粒状物用注射用水洗涤3次以除去表面活性剂。然后将洗涤过的颗粒状物在合适的容器中用注射用水重悬,随后冷冻干燥成包封第一化合物的粉末状微粒。然后通过以25kgy的最小剂量在干冰上进行γ照射,最终对微粒进行最终灭菌。微粒a:(药物:resomer752s1:1)微粒b:(药物:resomer752s1:2)。在研究当天,通过涡旋将微粒a(第一化合物:resomer752s1:1)和b(第一化合物:resomer752s1:2)分别与0.912ml和0.945ml载体混合,直到得到没有大的附聚物的视觉均匀的悬浮液。将药物悬浮液配制成均匀的1ml悬浮液,并通过涡旋重悬,直到获得目视均匀的悬浮液,没有大的附聚物。对于第一化合物的肌肉内施用途径,每个制剂给药三只雄性crl:cd大鼠并取样。第一化合物的肌内剂量以20mg/kg的单次剂量和0.5ml/kg的剂量体积施用。在剂量施用后的0.5、1、2、4、6、8、12和24小时和最高至1680小时收集血样。对于给药后的每个时间点,通过剪尾法收集约0.1ml血样,并立即冷冻并在-70℃储存直到分析。然后使用基于蛋白质沉淀的方法,之后通过lc-ms/ms分析,分析大鼠血样的第一化合物的浓度。该实施例的结果示于表3中并图示在图3中。表3途径(剂量)配方t1/2(天)cmax(ng/ml)tmax(hr)auc0-1680h/70天(h*mg/ml)im5mg/kg药物悬浮液16±0.6284.0±129.85.3±2.356.5±18.7im5mg/kg微粒a11±5.7612.7±143.48.7±3.149.5±7.8im5mg/ig微粒b10.0±3.5424.7±82.410.0±3.546.2±5.3当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1