清骨散颗粒的制备方法及质量控制方法与流程
本发明属于中药制剂领域,涉及清骨散颗粒的制备方法,还涉及清骨散颗粒质量控制方法。
背景技术:
清骨散出自于《证治准绳》,由柴胡、秦艽、胡黄连、鳖甲(醋制)、地骨皮、青蒿、知母、甘草8味药组成,具有清虚热退骨蒸的功效,主治阴虚发热,虚劳骨蒸。虽目前多用于治疗结核病,或其他慢性消耗性疾病的发热骨蒸属阴虚发热者,但外科临床亦辄有效验。方中银柴胡味甘,性微寒,直入阴分而清热凉血,善退虚劳骨蒸之热而无苦燥之弊,为君药。知母泻火滋阴退虚热;胡黄连入血分而清虚热;地骨皮凉血而退有汗之骨蒸,三药俱入阴退虚火,以助银柴胡清骨蒸潮热,共为臣药。秦艽、青蒿皆辛散透热之品,清虚热并透伏热使从外解;鳖甲咸寒,既滋阴潜阳,又引药入阴分,为治虚热之常用药,同为佐药。使以甘草,调和诸药,并防苦寒药物损伤胃气。本方重在清透伏热以治标,兼顾滋养阴液以治本,并收退热除蒸之效。
经典名方标准颗粒仿照传统中药汤剂的煎煮模式,既保留传统汤剂有效成分的特点,又避免了中成药难以随证加减和传统汤剂煎煮、携带不便的问题。该品种的开发,可形成“以经方标准颗粒为主,单味配方颗粒为辅,随证加减”的临床调剂模式,满足现代临床用药需求。本品的开发可以解决单味配方颗粒存在的无法共煎等争议的问题,充分发挥传统方剂配伍的优势,达到减毒增效的效果,尤为重要。复方配方颗粒不仅传承了中药单味配方颗粒的优点,同时又考虑了饮片在煎煮过程中的相互作用,符合中医传统用药理论,具有较大的价值。
由于我国尚无适合于临床调剂的复方颗粒,目前国际市场销售的中药复方颗粒均来自日本、韩国及我国台湾地区。该品种的研究将推进中药颗粒剂的国际市场开拓,促进中药国际化。
另外,由于目前还未见有对经典方清骨散颗粒的研究,对该品种的化学成分等方面的研究仍属空白,行业内缺少现行的质量标准,因此有必要研究一种完善的质量标准,以保证产品的安全有效、质量可控。
技术实现要素:
有鉴于此,本发明的目的之一在于提供一种经典方清骨散颗粒的制备方法,该方法能充分发挥中药配伍的特色及传统汤剂的优势,最大限度的保留了有效成分;本发明的目的之二在于提供清骨散颗粒的质量控制方法,建立了完善的质量标准,采用高效液相色谱法进行指纹图谱与多指标含量的质量控制,可有效控制复方颗粒的质量。
为达到上述目的,本发明提供如下技术方案:
清骨散颗粒的制备方法,包括如下步骤:
(1)按质量比为1-3:1-3:1-3:1-3:1-3:1-3:1-3:1-3分别取银柴胡、秦艽、胡黄连、醋制鳖甲、地骨皮、青蒿、知母、甘草,加水煎煮提取两次,合并煎煮液;
(2)将步骤(1)所得煎煮液,滤过,滤液真空减压浓缩至相对密度为1.16~1.22的浓缩液;
(3)将糊精和乳糖的混合物作为底物,喷入步骤(2)所得浓缩液中,沸腾干燥制粒,筛选粒度在16-60目的颗粒,得清骨散颗粒。
本发明步骤(1)中,所述银柴胡、秦艽、胡黄连、醋制鳖甲、地骨皮、青蒿、知母和甘草的质量比为3:2:2:2:2:2:2:1。
本发明步骤(1)中,所述煎煮提取两次具体为先加入相当于药材总重量5~11倍的水,煎煮提取1~3小时,过滤收集煎煮液,滤渣再加相当于药材总重量3~9倍的水,煎煮提取0.5~1.5小时,过滤,收集煎煮液。
本发明中,所述煎煮提取两次具体为先加入相当于药材总重量8倍的水,煎煮提取2小时,过滤收集煎煮液,滤渣再加相当于药材总重量6倍的水,煎煮提取1小时,过滤,收集煎煮液。
本发明步骤(3)中,所述糊精和乳糖的质量比为3﹕1。
本发明步骤(3)中,所述糊精和乳糖的混合物加入量为干膏量的1.31倍。
本发明步骤(3)中,所述沸腾干燥制粒的工艺参数为:控制进风温度:80℃、风机频率35hz、进料速度40r·min-1、雾化压力0.1mpa。
2、清骨散颗粒的质量控制方法,包括如下步骤:
(1)样品制备:将清骨散颗粒用体积分数为80%的甲醇溶液溶,超声提取30min,,冷却,再加体积分数为80%的甲醇溶液补足失重,摇匀,滤过,制得清骨散颗粒样品;
(2)对照品制备:分别取胆苦苷、胡黄连苷ⅱ和胡黄连苷ⅰ,用体积分数为80%的甲醇溶液制成含0.232mg/ml龙胆苦苷、0.269mg/ml胡黄连苷ⅱ和0.083mg/ml胡黄连苷ⅰ的混合溶液;
(3)色谱检测:将清骨散颗粒样品和对照品在如下条件下检测,色谱柱:agilentzorbaxsb-aq;柱温:25℃;进样量:10μl;流速:1.0ml/min;流动相:乙腈与磷酸质量分数为0.1%的磷酸水梯度洗脱;检测波长:210nm;
所述梯度洗脱条件为:0~15min,乙腈:磷酸质量分数为0.1%的磷酸水的体积比由5﹕95到15﹕85;35~50min,乙腈:磷酸质量分数为0.1%的磷酸水的体积比由15﹕85到18﹕82;50~65min,乙腈:磷酸质量分数为0.1%的磷酸水的体积比由30﹕70到55﹕45;65~75min,乙腈:磷酸质量分数为0.1%的磷酸水的体积比为55:45。
进一步,步骤(1)中所述甲醇溶液加入量为按每g清骨散颗粒加入50ml体积分数为80%的甲醇溶液。
本发明中,干膏量为中药经水煎煮,浓缩后干燥得到的膏体。
本发明的有益效果在于:本发明公开了清骨散颗粒的制备方法,解决了我国尚无适合于临床调剂的复方颗粒,本发明首次公开了经典名方清骨散标准颗粒的制备工艺,另外,对经典方清骨散颗粒的成分进行研究,提供了一种多指标含量测定控制产品的方法,解决了清骨散颗粒在化学成分等方面的研究空白,弥补了行业内缺少的质量标准,以保证产品的安全有效、质量可控。
附图说明
为了使本发明的目的、技术方案和有益效果更加清楚,本发明提供如下附图进行说明:
图1为实施例4的不同色谱柱的hplc色谱图(1:龙胆苦苷;2:胡黄连苷ⅱ;3:胡黄连苷ⅰ;a:agilentzorbaxsb-aq;b:phenomenexluna5uc18(2);c:kromasil100-5-c18)。
图2为实施例4的不同流动相系统的考察的hplc色谱图(1:龙胆苦苷;2:胡黄连苷ⅱ;3:胡黄连苷ⅰ;a:乙腈-0.1%磷酸水;b:乙腈-0.3%磷酸;c:乙腈-水)。
图3为实施例4的不同流速的考察的hplc色谱图(1:龙胆苦苷;2:胡黄连苷ⅱ;3:胡黄连苷ⅰ;a:流速0.8ml/min;b:流速1.0ml/min;c:流速1.2ml/min)。
图4为实施例4的不同柱温的考察的hplc色谱图(1:龙胆苦苷;2:胡黄连苷ⅱ;3:胡黄连苷ⅰ;a:柱温25℃;b:柱温30℃;c:柱温35℃)。
图5实施例4的龙胆苦苷标准曲线图。
图6实施例4的胡黄连苷ⅱ标准曲线图。
图7实施例4的胡黄连苷ⅰ标准曲线图。
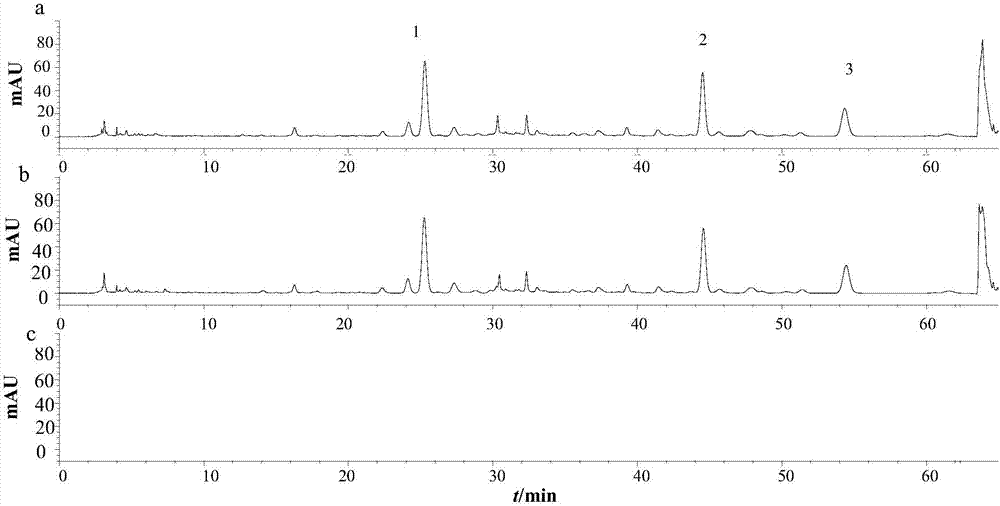
图8实施例4的专属性试验色谱图(1:龙胆苦苷;2:胡黄连苷ⅱ;3:胡黄连苷ⅰ;a:清骨散颗粒;b:秦艽-阴性;c:胡黄连-阴性;d:糊精-乳糖)。
图9为清骨散颗粒的标准hplc指纹图谱。
具体实施方式
下面将结合附图,对本发明的优选实施例进行详细的描述。
实施例1
清骨散颗粒的制备方法,包括如下步骤:
a、取银柴胡300g,秦艽200g,胡黄连200g,鳖甲(醋制)200g,地骨皮200g,青蒿200g,知母200g,甘草100g,加12.8kg的水,煎煮提取2.0小时,即一煎;一煎后药渣加入9.6kg的水,煎煮提取1.0小时,即二煎;
b、合并两煎滤液,滤过,滤液真空减压浓缩至相对密度为1.18~1.19(60℃)的浓缩液;
c、加入重量比为3:1的糊精及乳糖混合物混匀,作为底物,喷入上述浓缩液,沸腾干燥制粒,控制进风温度:80℃、风机频率35hz、进料速度40r·min-1、雾化压力0.1mpa,筛选粒度在16-60目的颗粒,得清骨散颗粒。
实施例2
清骨散颗粒的制备方法,包括如下步骤:
a、取银柴胡300g,秦艽200g,胡黄连200g,鳖甲(醋制)200g,地骨皮200g,青蒿200g知母200g,甘草100g,加16kg的水,煎煮提取2.5小时,即一煎;一煎后药渣加入12.8kg的水,煎煮提取1.5小时,即二煎;
b、合并两煎滤液,滤过,滤液真空减压浓缩至相对密度为1.19~1.20(60℃)的浓缩液;
c、加入重量比为3:1的糊精及乳糖的混合物混匀,作为底物,喷入上述浓缩液,沸腾干燥制粒,控制进风温度:80℃、风机频率35hz、进料速度40r·min-1、雾化压力0.1mpa,筛选粒度在16-60目的颗粒,得清骨散颗粒。
实施例3
清骨散颗粒的制备方法,包括如下步骤:
a、取银柴胡300g,秦艽200g,胡黄连200g,鳖甲(醋制)200g,地骨皮200g,青蒿200g知母200g,甘草100g,加9.6kg的水,煎煮提取1.5小时,即一煎;一煎后药渣加入6.4kg的水,煎煮提取0.5小时,即二煎;
b、合并两煎滤液,滤过,滤液真空减压浓缩至相对密度为1.20~1.21(60℃)的浓缩液;
c、加入重量比为3:1的糊精及乳糖混合物混匀,作为底物,喷入上述浓缩液,沸腾干燥制粒,控制进风温度:80℃、风机频率35hz、进料速度40r·min-1、雾化压力0.1mpa,筛选粒度在16-60目的颗粒,得清骨散颗粒。
实施例4
清骨散颗粒的制备方法,包括步骤如下:
a、取银柴胡300g,秦艽200g,胡黄连200g,鳖甲(醋制)200g,地骨皮200g,青蒿200g知母200g,甘草100g,加12.8kg的水,煎煮提取2.0小时,即一煎;一煎后药渣加入9.6kg的水,煎煮提取1.0小时,即二煎;
b、合并两煎滤液,滤过,滤液真空减压浓缩至相对密度为1.18~1.20(60℃)的浓缩液;
c、加入重量比为3:1的糊精及乳糖混合物物混匀,作为底物,喷入上述浓缩液,沸腾干燥制粒,控制进风温度:80℃、风机频率35hz、进料速度40r·min-1、雾化压力0.1mpa,筛选粒度在16-60目的颗粒,得清骨散颗粒。
实施例5、清骨散颗粒进行含量测定
制备后,采用高效液相色谱法对清骨散颗粒进行含量测定,具体步骤如下:
(1)实验材料与仪器:
实验材料:龙胆苦苷对照品:中国药品生物制品检定所,批号110770-201515;胡黄连苷ⅱ对照品:中国药品生物制品检定所,批号111596-201103;胡黄连苷ⅰ对照品:中国药品生物制品检定所,批号111727-200501;乙腈:色谱纯,天津市彪仕奇科技发展有限公司;甲醇:色谱纯,天津市大茂化学试剂厂产品;水:超纯水;清骨散颗粒(太极集团重庆涪陵制药厂有限公司生产)。
仪器:高效液相色谱仪:agilent1100高效液相色谱系统;紫外检测器:vwdg1314a;sartorius电子天平:bs2202smax2200gd=0.01g;sartorius电子天平:bs124smax120gd=0.0001g;sartorius电子天平:bt25smax2200gd=0.01mg;kh5200型超声波清洗器,昆山禾创超声仪器有限公司:色谱柱:agilentzorbaxsb-aq4.6×250mm5μm。
(2)供试品溶液的制备方法
结合秦艽及胡黄连药材的成分特点,本实验对供试品溶液的制备方法进行了考察,计算指标性成分龙胆苦苷、胡黄连苷ⅱ和胡黄连苷ⅰ的含量。
a.提取溶剂的考察
由于本品是水提取物,亲水物质占主要成分,故在含量测定方法确立时重新摸索提取溶剂,纯甲醇渗透性较好,首选甲醇为提取溶剂,结合文献和药典方法,同时考虑水、稀乙醇、50%甲醇和80%甲醇。结果见表1。
表1、提取溶剂的考察—龙胆苦苷、胡黄连苷ⅱ和胡黄连苷ⅰ含量测定结果
结果表明,用80%甲醇作为提取溶剂,龙胆苦苷、胡黄连苷ⅱ和胡黄连苷ⅰ含量均较高。
b.提取方法的考察
拟采用超声、回流和振摇作为提取方法进行了考察。方法为:取装量差异项下样品,研细,取1.0g,精密称定,置具塞锥形瓶中,6份,分别精密加入80%甲醇50ml,称定重量,分别按上述3种提取方法提取,时间30min,每种方法平行两份。计算龙胆苦苷、胡黄连苷ⅱ和胡黄连苷ⅰ的含量,结果见表2。
表2、提取方法的考察
结果表明,超声及回流对龙胆苦苷、胡黄连苷ⅱ和胡黄连苷ⅰ的提取效果较好,考虑操作方便,选择超声作为清骨散颗粒的提取方法。
c.提取时间的考察
拟对不同的提取时间15min,30min,60min进行了考察。方法为:取装量差异项下样品,研细,取1.0g,精密称定,置具塞锥形瓶中,6份,分别精密加入80%甲醇50ml,称定重量,分别按上述3种提取时间超声提取,每种提取时间平行两份。计算龙胆苦苷、胡黄连苷ⅱ和胡黄连苷ⅰ的含量,结果见表3。
表3、提取时间的考察
结果表明,3种超声时间对龙胆苦苷、胡黄连苷ⅱ和胡黄连苷ⅰ提取效果相差不多,为保证提取完全,选择超声时间为30min。
d.溶剂量的考察
拟对溶剂不同的体积25ml、50ml、100ml进行了考察。方法为:取装量差异项下样品,研细,取1.0g,精密称定,置具塞锥形瓶中,6份,分别精密加入80%甲醇25ml、50ml、100ml,称定重量,超声提取30min,平行两份。计算龙胆苦苷、胡黄连苷ⅱ和胡黄连苷ⅰ的含量,结果见表4。
表4、溶剂量的考察
结果表明,溶剂量为50ml和100ml对龙胆苦苷、胡黄连苷ⅱ和胡黄连苷ⅰ的提取效果较好,且溶剂量为50ml时能将龙胆苦苷、胡黄连苷ⅱ和胡黄连苷ⅰ提取完全。
e、确定供试品溶液的制备方法
取装量差异项下颗粒样品,研细,取1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇50ml,称定重量,超声处理(功率200w,频率40khz)30min,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
(3)色谱条件与系统适用性试验
a.色谱柱的考察
考察了agilentzorbaxsb-aq、phenomenexluna5uc18(2)及kromasil100-5-c183种色谱柱,结果如附图1,结果以agilentzorbaxsb-aq色谱柱分离效果最好。
b.流动相的考察
考察了乙腈-0.1%磷酸水、乙腈-0.3%磷酸水和乙腈-水3种流动相系统,结果如附图2。结果以乙腈-0.1%磷酸水流动相分离效果好。
c.流速的选择
分别考察流速:0.8ml/min、1.0ml/min、1.2ml/min,结果以流速1.0ml/min分离效果良好,可将3个色谱峰完全分离。见附图3。
d.柱温的选择
分别考察柱温:25度、30度、35度,结果以柱温25度分离效果良好,可将3个色谱峰完全分离。见附图4。
(4)方法学考察
a.标准曲线与线性范围
取龙胆苦苷、胡黄连苷ⅱ和胡黄连苷ⅰ对照品适量,精密称定,加80%甲醇制成每1ml含龙胆苦苷0.232mg、胡黄连苷ⅱ0.269mg和胡黄连苷ⅰ0.083mg的混合溶液,即得。精密吸取对照品溶液1、2、5、10、12、15μl进样,记录峰面积,以进样量(ug)为横坐标,峰面积为纵坐标绘制标准曲线(见附图5、6和7),测定结果见表5。
龙胆苦苷:y=1335.6x+2.8898r2=1
胡黄连苷ⅱ:y=980.1x+7.1551r2=0.9997
胡黄连苷ⅰ:y=2324.3x+1.7362r2=1
结果表明:龙胆苦苷对照品、胡黄连苷ⅱ对照品和胡黄连苷ⅰ对照品分别在0.232~3.478μg,0.269~4.035μg,0.083~1.245μg范围内呈良好的线性关系。
表5、龙胆苦苷、胡黄连苷ⅱ和胡黄连苷ⅰ对照品标准曲线结果
b.精密度考察取颗粒(批号:16050002),研细,按供试品溶液的制备方法制备供试品,连续进样6次,测定峰面积,计算rsd值,结果见表6,结果表明,仪器精密度良好。
表6、精密度试验结果
c.稳定性考察取颗粒(批号:16050002),研细,按供试品溶液的制备方法制备供试品,分别在0、3、6、9、12、24小时进样,结果见表7。结果表明,样品溶液在24小时内测定能够满足含量测定的误差要求。
表7、样品稳定性试验结果
d.重复性试验取颗粒(批号为16050002),研细,按供试品溶液的制备方法制备供试品,平行6份,测定峰面积,计算含量。结果见表8。结果表明,方法的重复性较好。
表8、重复性试验结果
e.准确性试验即加样回收试验,选取已知含量的颗粒(批号为16050002),(龙胆苦苷含量为9.96mg/g,胡黄连苷ⅱ含量为11.19mg/g,胡黄连苷ⅰ含量为3.04mg/g),研细,精密量取样品0.25g,按1︰1浓度分别精密加入对照品溶液,按供试品溶液的制备方法,制成6份同浓度的供试品溶液。按【含量测定】方法测定,计算出回收率,见表9~11。结果表明,
方法准确性符合含量测定的要求。
表9、龙胆苦苷—加样回收率试验结果
表10、胡黄连苷ⅱ—加样回收率试验结果
表11、胡黄连苷ⅰ—加样回收率试验结果
f.专属性试验
取清骨散颗粒除去秦艽药材、除去胡黄连药材及糊精-乳糖的阴性对照样品,研细,分别取0.5g,按上文中供试品溶液的制备方法制备,按【含量测定】项下所述的色谱条件进行hplc分析,结果表明清骨散颗粒的阴性对照对其含量测定无干扰,该含量测定方法具有一定的专属性。结果见附图8。
(5)清骨散颗粒中秦艽、胡黄连的含量测定
参照正文的方法,对3批清骨散颗粒中秦艽、胡黄连的含量进行了测定,结果见表12。
表12、3批清骨散颗粒中龙胆苦苷、胡黄连苷ⅱ和胡黄连苷ⅰ的含量测定结果
故暂规定本品每袋含龙胆苦苷不得少于30.28mg,含胡黄连苷ⅱ不得少于35.49mg,胡黄连苷ⅰ不得少于8.98mg。
(6)清骨散颗粒的指纹图谱研究
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.1%磷酸水为流动相b,按下表13条件进行梯度洗脱;流速1.0ml/min;柱温:35℃;检测波长275nm;理论塔板数按龙胆苦苷峰计算应不低于3000。
表13、梯度洗脱条件
供试品溶液的制备取实施例4的秦艽、胡黄连的供试品溶液。
对照品溶液的制备取实施例4的秦艽、胡黄连的对照品溶液。
测定法分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,记录75分钟的色谱图(图9),即得。
供试品指纹图谱与对照指纹图谱应在相应保留时间处出现相同色谱峰,两者相似度应不低于0.90。
最后说明的是,以上优选实施例仅用以说明本发明的技术方案而非限制,尽管通过上述优选实施例已经对本发明进行了详细的描述,但本领域技术人员应当理解,可以在形式上和细节上对其作出各种各样的改变,而不偏离本发明权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!