杂环化合物和其用途的制作方法
相关申请的交叉引用
本申请要求2014年3月14日提交的美国临时申请第61/785,763号的权益,所述申请的公开内容以全文引用的方式并入本文中。
领域
本发明提供包含奥美卡替莫卡必尔(omecamtivmecarbil)或其药学上可接受的盐、药学上可接受的水合物或药学上可接受的盐的药学上可接受的水合物的药物制剂,如奥美卡替莫卡必尔二盐酸盐水合物。
背景
心脏肌小节为心脏中肌肉收缩的基本单位。心脏肌小节为由心肌肌凝蛋白、肌动蛋白和一组调节蛋白构成的高度有序细胞骨架结构。小分子心肌肌凝蛋白活化剂的发现与发展将导向对急性及慢性心脏衰竭的有前景的治疗。心肌肌凝蛋白为心肌细胞中的细胞骨架动力蛋白。它直接负责将化学能转化成机械力,从而导致心肌收缩。
当前正性心肌收缩剂,如β-肾上腺素能受体激动剂或磷酸二酯酶活性抑制剂,增加细胞内钙的浓度,借此增加心脏肌小节收缩性。然而,钙含量增加会增加心肌收缩速度并缩短收缩期射血时间,这已与潜在威胁生命的副作用相关。相比之下,心肌肌凝蛋白活化剂通过直接刺激心肌肌凝蛋白动力蛋白活性的机制起作用,而不会增加细胞内钙浓度。它们加速肌凝蛋白酶循环的限速步骤并使其有利于力产生状态偏移。这种机制并非增加心脏收缩的速度,而是缩短收缩期射血时间,这以潜在更具氧气有效的方式增加心肌收缩性和心脏输出。
美国专利第7,507,735号(以引用的方式并入本文中)公开一种化合物,其包括奥美卡替莫卡必尔(amg423,ck-1827452),具有以下结构:
奥美卡替莫卡必尔为心脏肌凝蛋白(即引起心脏收缩的动力蛋白)的首创直接活化剂。它在静脉内和口服制剂中均作为潜在心脏衰竭治疗物来评估,目标为建立针对住院和门诊设置中的患者的新的持续护理。
提供奥美卡替莫卡必尔的i.v.递送的临床试验已展示这种药物的血浆含量可安全且有效地递送。然而,标准释放制剂和一些延长释放制剂给出的峰谷比可能过大,以致无法对慢性或预防设置中需要所述药物的患者提供安全且有效量的奥美卡替莫卡必尔(参见图4)。因此,将需要有效的持续释放制剂来达到增加的患者安全性和有效性。
概述
提供一种药物制剂,其包含:
奥美卡替莫卡必尔或其药学上可接受的盐、药学上可接受的水合物或药学上可接受的盐的药学上可接受的水合物;
控制释放剂;
ph调节剂;填充剂;以及
润滑剂。
本发明还提供一种用于制备药物制剂的方法,其包括:
共混包含奥美卡替莫卡必尔或其药学上可接受的盐、药学上可接受的水合物或药学上可接受的盐的药学上可接受的水合物、控制释放剂、ph调节剂以及填充剂的混合物;
使用润滑剂润滑共混的混合物;
将润滑的共混物造粒;
使用润滑剂润滑所得造粒物;并且
将润滑的造粒物压缩成所需的形式。
还提供一种治疗选自急性心脏衰竭和慢性心脏衰竭的疾病的方法,其包括向有需要的患者施用本文所述的药物制剂。
附图说明
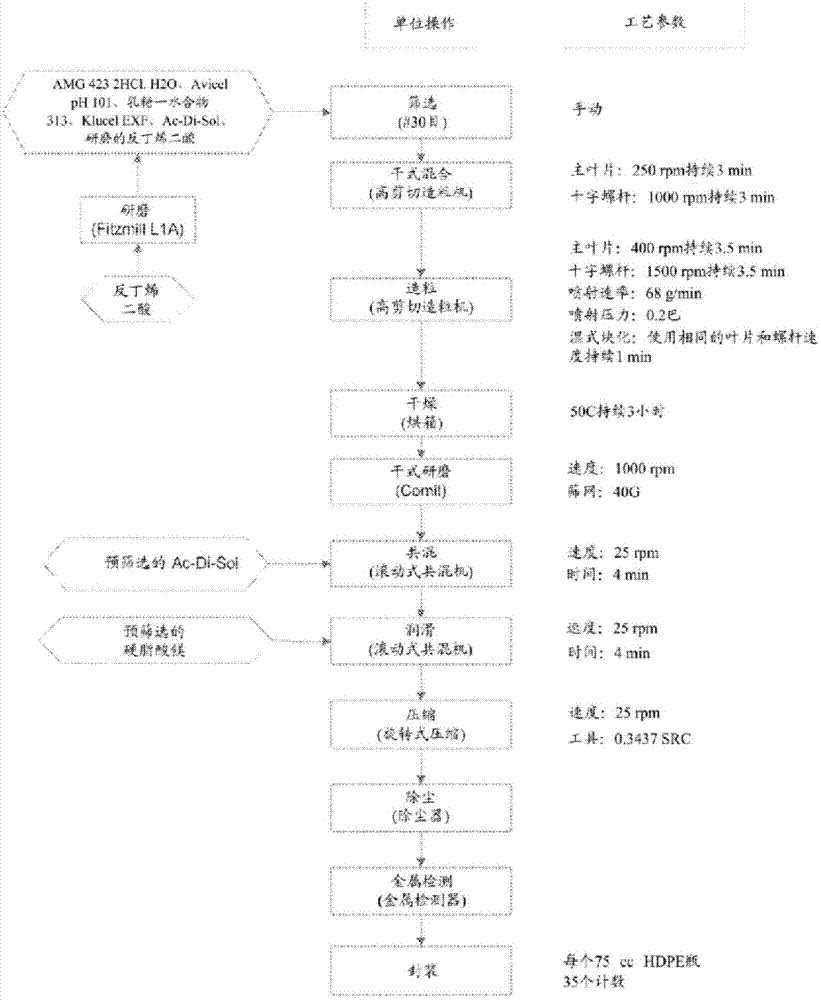
图1为制备奥美卡替莫卡必尔的立即释放(ir)片剂(25mg)的流程图;参见实施例1。
图2为制备基质改性的释放组合物的流程图;参见实施例2。
图3为制备基质改性的释放组合物的流程图;参见实施例3-5。
图4展示针对一种立即释放组合物(ir)和两种基质改性的释放组合物(mtx-f1和mtx-f2),在禁食(顶部)和进食(底部)的情况下,健康自愿者的暴露量(血浆浓度(ng/ml)对时间(h));研究是在健康成人受试者中进行的随机化、开放标签、4路交叉不完全分块设计研究:
●60位受试者;1个场地,美国
●总共12次治疗(每次治疗耗时20分钟)
●各种制剂;禁食或进食情况下服用每种制剂
●每位受试者将被随机分到1个序列
●每位受试者接收12种可能治疗中的4种
●每个周期约为7天;研究持续时间:27天(周期4:5天)。
图5为具有针对一种立即释放组合物(ir)和两种基质改性的释放组合物(mtx-f1及mtx-f2)的数据的表。
图6展示在两种ph(2和6.8)下针对一种奥美卡替莫卡必尔游离碱的基质制剂(顶部)和针对一种奥美卡替莫卡必尔二盐酸盐水合物盐形式即形式a(底部)的药物释放。
图7示出形式a的x射线粉末衍射图(xrpd)。
图8展示在变化的相对湿度条件下,奥美卡替莫卡必尔二盐酸盐水合物盐形式的xrpd。
图9展示在变化的温度下,奥美卡替莫卡必尔二盐酸盐水合物盐形式的xrpd。
图10展示奥美卡替莫卡必尔二盐酸盐的形式a、b及c的xrpd图样的重叠图。
详述
除非另外规定,否则以下定义适用于本说明书和权利要求书中可见的术语:
“治疗”或“处理”指对患者疾病的任何治疗,包括:a)预防疾病,即,致使疾病的临床症状不再发展;b)抑制疾病;c)减慢或停止临床症状的发展;和/或d)缓解疾病,即,临床症状产生消退。本文的疾病和病症的治疗也意图包括向被认为需要预防性治疗(例如像慢性心脏衰竭)的受试者(即动物,优选哺乳动物、最优选人)进行本文所描述的药物制剂的预防施用。
术语“治疗有效量”指的是当向人或非人患者施用时,对治疗疾病有效的量,例如治疗有效量可以是足以治疗响应于肌凝蛋白活化的疾病或病症的量。治疗有效量可以实验方法确定,例如通过分析化学实体的血液浓度确定,或在理论上通过计算生物有效性确定。
“药学上可接受的盐”包括但不限于与无机酸所成的盐,如盐酸盐(即氢氯化物)、磷酸盐、二磷酸盐、氢溴酸盐、硫酸盐、亚磺酸盐、硝酸盐和类似盐;以及与有机酸所成的盐,如苹果酸盐、马来酸盐、反丁烯二酸盐、酒石酸盐、琥珀酸盐、柠檬酸盐、乙酸盐、乳酸盐、甲磺酸盐、对甲苯磺酸盐、2-羟乙基磺酸盐、苯甲酸盐、水杨酸盐、硬脂酸盐,和链烷酸盐,如乙酸盐、hooc--(ch2)n--cooh(其中n是0-4)和类似盐。同样地,药学上可接受的阳离子包括但不限于钠、钾、钙、铝、锂和铵。本领域的技术人员将会认识到可用于制备无毒的药学上可接受的加成盐的各种合成方法。
术语“水合物”指的是通过水和化合物的相互作用形成的化学实体,包括例如半水合物、一水合物、二水合物、三水合物等。
“结晶形式”、“多晶型物”以及“新形式”在本文可互换使用并且意图包括化合物的所有的结晶以及无定形形式,包括例如多晶型物、假多晶型物、溶剂化物、水合物、未溶剂化的多晶型物(包括无水化合物)、构象多晶型物和无定形形式以及其混合物,除非指出具体的结晶或无定形形式。
本说明书和权利要求书含有使用语言“选自……和……”和“为……或……”的物种清单(有时称为马库什组)。当这种语言用于本申请中时,除非另外陈述,否则其意图包括群组整体,或其任何单一成员,或其任何子组。这种语言的使用仅仅用于简写目的且不意图以任何方式限制如所需的个别要素或子组的移除。
本发明提供包含奥美卡替莫卡必尔或其药学上可接受的盐、药学上可接受的水合物或药学上可接受的盐的药学上可接受的水合物的药物制剂,如奥美卡替莫卡必尔二盐酸盐水合物。
本文所描述的药物制剂能够释放奥美卡替莫卡必尔,甚至通过奥美卡替莫卡必尔穿过由片剂中的控制释放剂的水合作用形成的凝胶层的扩散受控的均匀的释放。在一些实施方案中,与其他上文或下文实施方案结合,本发明的改性释放基质片剂证明在体外的最小ph依赖性释放。在一些实施方案中,与其他上文或下文实施方案结合,奥美卡替莫卡必尔在24小时内在ph为2以及6.8的溶解介质中都实现完全释放,可能导致更小的受试者间和受试者内的可变性以及食物影响。据发现,本发明的改性释放基质片剂剂型优于以前的立即释放剂型,使血浆峰谷比减到最小。结果,本发明的改性释放基质片剂减小血浆浓度导致减小的副作用以及改进的安全性和效力。也预期本发明的改性释放基质片剂将通过减少给药频率改善患者的依从性。此外,本发明的改性释放基质片剂是物理化学上稳定的——导致其在40℃/75%的相对湿度(rh)下存储6个月后物理属性、分析、杂质或溶解曲线都没有变化。
在一些实施方案中,与其他上文或下文实施方案结合,在人体内给药后2至12小时,奥美卡替莫卡必尔的暴露量是在40ng/ml与70ng/ml之间。
在一些实施方案中,与其他上文或下文实施方案结合,在人体内给药后2至12小时,奥美卡替莫卡必尔的暴露量保持在40ng/ml与55ng/ml之间。
在一些实施方案中,与其他上文或下文实施方案结合,所述奥美卡替莫卡必尔在以下间隔释放:
在1小时时≤30%溶解剂量;
在1小时时30-75%溶解剂量;以及
在12小时时≥80%溶解剂量。
在一些实施方案中,与其他上文或下文实施方案结合,所述奥美卡替莫卡必尔在以下间隔释放:
在2小时时≤30%溶解剂量;
在6小时时30-75%溶解剂量;以及
在16小时时≥80%溶解剂量。
提供药物制剂,其包含:
奥美卡替莫卡必尔或其药学上可接受的盐、药学上可接受的水合物或药学上可接受的盐的药学上可接受的水合物;
控制释放剂;
ph调节剂;
填充剂;以及
润滑剂。
在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含约
3-30%w/w的奥美卡替莫卡必尔或其药学上可接受的盐、药学上可接受的水合物或药学上可接受的盐的药学上可接受的水合物;
15-35%w/w的控制释放剂;
20-45%w/wph的调节剂;
25-65%w/w的填充剂;以及
0.1-1.0%w/w的润滑剂。
在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含约
12-25(w/w%)奥美卡替莫卡必尔二盐酸盐水合物;25-35(w/w%)methoceltmk100mpremcr;20-30(w/w%)微晶纤维素,ph102;5-10(w/w%)乳糖一水合物,ff316;12-25(w/w%)反丁烯二酸;0.1-2(w/w%)粒内硬脂酸镁;以及0.1-2(w/w%)粒外硬脂酸镁。
在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含约:
3-10(w/w%)奥美卡替莫卡必尔二盐酸盐水合物;20-40(w/w%)methoceltmk100mpremcr;30-42(w/w%)微晶纤维素,ph102;12-25(w/w%)乳糖一水合物,ff316;4-11(w/w%)反丁烯二酸;0.1-2(w/w%)粒内硬脂酸镁;以及0.1-2(w/w%)粒外硬脂酸镁。
在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含约:
12-25(w/w%)奥美卡替莫卡必尔二盐酸盐水合物;1-10(w/w%)methoceltmk100mpremcr;12-27(w/w%)methoceltmk100lvpremcr;20-35(w/w%)微晶纤维素,ph102;4-15(w/w%)乳糖一水合物,ff316;12-25(w/w%)反丁烯二酸;0.1-2(w/w%)粒内硬脂酸镁;以及0.1-2(w/w%)粒外硬脂酸镁。
在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含约:
3-10(w/w%)奥美卡替莫卡必尔二盐酸盐水合物;1-10(w/w%)methoceltmk100mpremcr;12-27(w/w%)methoceltmk100lvpremcr;30-50(w/w%)微晶纤维素,ph102;15-25(w/w%)乳糖一水合物,ff316;3-11(w/w%)反丁烯二酸;0.1-2(w/w%)粒内硬脂酸镁;以及0.1-2(w/w%)粒外硬脂酸镁。
在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含约:
18-19(w/w%)奥美卡替莫卡必尔二盐酸盐水合物;28-32(w/w%)methoceltmk100mpremcr;23-26(w/w%)微晶纤维素,ph102;7-9(w/w%)乳糖一水合物,ff316;17-20(w/w%)反丁烯二酸;0.1-1(w/w%)粒内硬脂酸镁;以及0.1-1(w/w%)粒外硬脂酸镁。
在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含约:
5-7(w/w%)奥美卡替莫卡必尔二盐酸盐水合物;27-33(w/w%)methoceltmk100mpremcr;35-38(w/w%)微晶纤维素,ph102;17-20(w/w%)乳糖一水合物,ff316;6-9(w/w%)反丁烯二酸;0.1-1(w/w%)粒内硬脂酸镁;以及0.1-1(w/w%)粒外硬脂酸镁。
在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含约:
17-20(w/w%)奥美卡替莫卡必尔二盐酸盐水合物;3-7(w/w%)methoceltmk100mpremcr;18-22(w/w%)methoceltmk100lvpremcr;26-30(w/w%)微晶纤维素,ph102;8-11(w/w%)乳糖一水合物,ff316;17-20(w/w%)反丁烯二酸;0.1-1(w/w%)粒内硬脂酸镁;以及0.1-1(w/w%)粒外硬脂酸镁。
在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含约:
5-7(w/w%)奥美卡替莫卡必尔二盐酸盐水合物;3-7(w/w%)methoceltmk100mpremcr;18-22(w/w%)methoceltmk100lvpremcr;37-43(w/w%)微晶纤维素,ph102;18-22(w/w%)乳糖一水合物,ff316;6-9(w/w%)反丁烯二酸;0.1-1(w/w%)粒内硬脂酸镁;以及0.1-1(w/w%)粒外硬脂酸镁。
在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含约:
18.37(w/w%)奥美卡替莫卡必尔二盐酸盐水合物;30(w/w%)methoceltmk100mpremcr;24.20(w/w%)微晶纤维素,ph102;8.07(w/w%)乳糖一水合物,ff316;18.37(w/w%)反丁烯二酸;0.5(w/w%)粒内硬脂酸镁;以及0.5(w/w%)粒外硬脂酸镁。
在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含约:
6.13(w/w%)奥美卡替莫卡必尔二盐酸盐水合物;30(w/w%)methoceltmk100mpremcr;36.81(w/w%)微晶纤维素,ph102;18.40(w/w%)乳糖一水合物,ff316;7.66(w/w%)反丁烯二酸;0.5(w/w%)粒内硬脂酸镁;以及0.5(w/w%)粒外硬脂酸镁。
在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含约:
18.37(w/w%)奥美卡替莫卡必尔二盐酸盐水合物;5(w/w%)methoceltmk100mpremcr;20(w/w%)methoceltmk100lvpremcr;27.95(w/w%)微晶纤维素,ph102;9.31(w/w%)乳糖一水合物,ff316;18.37(w/w%)反丁烯二酸;0.5(w/w%)粒内硬脂酸镁;以及0.5(w/w%)粒外硬脂酸镁。
在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含约:
6.13(w/w%)奥美卡替莫卡必尔二盐酸盐水合物;5(w/w%)methoceltmk100mpremcr;20(w/w%)methoceltmk100lvpremcr;40.14(w/w%)微晶纤维素,ph102;20.07(w/w%)乳糖一水合物,ff316;7.66(w/w%)反丁烯二酸;0.5(w/w%)粒内硬脂酸镁;以及0.5(w/w%)粒外硬脂酸镁。
奥美卡替莫卡必尔
在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含奥美卡替莫卡必尔二盐酸盐。在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含奥美卡替莫卡必尔二盐酸盐水合物。在一些实施方案中,与其他上文或下文实施方案结合,所述药物制剂包含奥美卡替莫卡必尔二盐酸盐水合物形式a。
在一些实施方案中,与其他上文或下文实施方案结合,形式a可通过x射线粉末衍射图样来表征,如实施例中所述而获得,使用cukα辐射时具有在约6.6、14.9、20.1、21.4和26.8±0.2°2θ处的峰。形式a任选地可通过x射线粉末衍射图样进一步表征,使用cukα辐射时具有在约8.4、24.2、26.0、33.3±0.2°2θ处的额外峰。形式a任选地可甚至通过x射线粉末衍射图样进一步表征,使用cukα辐射时具有在约6.2、9.7、13.2、14.3、15.4、16.3、16.9、18.9、19.5、20.7、21.8、22.8、23.6、25.1、27.3、27.7、28.4、29.4、30.2、31.2、31.5、31.9、33.9、34.5、34.9、36.1、36.8、37.7、38.5和39.7±0.2°2θ处的额外峰。在各种情形下,形式a可通过xrpd图样来表征,使用cukα辐射时具有在约6.2、6.6、8.4、9.7、13.2、14.3、14.9、15.4、16.3、16.9、18.9、19.5、20.1、20.7、21.4、21.8、22.8、23.6、24.3、25.1、26.0、26.8、27.3、27.7、28.4、29.4、30.2、31.2、31.5、31.9、33.3、33.9、34.5、34.9、36.1、36.8、37.7、38.5和39.7±0.2°2θ处的峰。在一些实施方案中,与其他上文或下文实施方案结合,形式a可通过实质上如图7所描述的x射线粉末衍射图样来表征,其中“实质上”意指所报告的峰可变化约±0.2°。在xrpd领域中众所周知,当光谱中的相对峰高依赖于多种因素,如样品制备以及仪器几何形状时,峰位置对于实验细节相对不敏感。
奥美卡替莫卡必尔的形式b和形式c多晶型物是亚稳态无水二盐酸盐形式,并且可在变化的水合条件下形成,如图8和图9中所注明的。特征形式b2-θ值包括使用cukα辐射时6.8、8.8、14.7、17.7和22.3±0.2°2θ,并且可另外包括使用cukα辐射时在9.6、13.5、19.2、26.2±0.2°2θ处的峰。形式b可以xrpd图样来表征,使用cukα辐射时具有在6.2、6.8、8.8、9.6、13.5、14.4、14.7、15.4、16.3、17.0、17.7、18.3、19.2、19.9、20.5、20.8、21.8、22.3、22.7、23.0、24.8、25.1、25.5、26.2、26.4、26.8、27.5、28.5、30.2、30.6、31.1、31.5、32.1、32.7、34.1、34.4、35.5、35.9、38.1、38.9±0.2°2θ处的峰。特征形式c2-θ值包括使用cukα辐射时6.7、14.8、17.4、20.6和26.2±0.2°2θ,并且可另外包括使用cukα辐射时在8.7、22.0、27.1和27.7±0.2°2θ处的峰。形式c可以xrpd图样来表征,使用cukα辐射时具有在6.2、6.7、8.7、9.6、13.5、14.5、14.8、15.4、16.4、17.1、17.4、18.4、19.3、19.5、19.9、20.6、20.8、21.8、22.0、22.5、22.8、24.3、24.7、25.1、25.6、26.2、26.5、27.1、27.3、27.7、28.5、30.0、30.5、31.0、31.5、32.2、32.8、34.1、35.2、36.0、36.9和38.8±0.2°2θ处的峰。
也参图9(变化的温度的xrpd数据)、图8(变化的相对湿度的xrpd数据)以及图10(重叠图)
控制释放剂
如本文所用,术语“控制释放剂”指的是促进活性成分以受控的方式从当前组合物释放的药剂。在一些实施方案中,与其他上文或下文实施方案结合,所述控制释放剂形成凝胶组合物。控制释放剂包括普鲁兰、糊精、钠及钙酸、聚丙烯酸、聚甲基丙烯酸、聚甲基乙烯醚共-顺丁烯二酸酐、聚乙烯吡咯烷酮、聚氧化乙烯、聚乙二醇、羟丙基纤维素、羟丙基甲基纤维素、羟基乙基纤维素、甲基丙烯酸羟基甲酯、羧基甲基纤维素钠、羧基甲基纤维素钙、甲基纤维素、麦芽糊精、三仙胶、黄蓍胶、琼脂、结冷胶、卡亚拉胶、褐藻酸、果胶、预胶凝化淀粉、聚乙烯醇、羧基甲基乙基纤维素、邻苯二甲酸乙酸纤维素、乙酸丁二酸纤维素、邻苯二甲酸甲基纤维素、邻苯二甲酸羟甲基乙基纤维素、邻苯二甲酸羟丙基甲基纤维素、乙酸丁二酸羟丙基甲基纤维素、聚乙烯醇邻苯二甲酸酯、聚丁酸乙烯邻苯二甲酸酯、聚乙烯缩醛邻苯二甲酸酯、乙酸乙烯酯/顺丁烯二酸酐共聚物、苯乙烯/顺丁烯二酸单酯共聚物、丙烯酸甲酯/甲基丙烯酸共聚物、苯乙烯/丙烯酸共聚物、丙烯酸甲酯/甲基丙烯酸/丙烯酸辛酯共聚物、甲基丙烯酸/甲基丙烯酸甲酯共聚物、苄基氨基甲基纤维素、二乙基氨基甲基纤维素、哌啶基乙基羟基乙基纤维素、乙酸二甲基氨基乙酸纤维素、乙烯基二乙胺/乙酸乙烯酯共聚物、乙烯基苄胺/乙酸乙烯酯共聚物、聚乙烯缩醛二乙基氨基乙酸酯、乙烯基哌啶基乙酰缩醛/乙酸乙烯酯共聚物、聚二乙基氨基甲基苯乙烯、甲基丙烯酸甲酯/甲基丙烯酸丁酯/甲基丙烯酸二甲基氨基乙酯和聚甲基丙烯酸二甲基氨基乙酯的共聚物、2-甲基-5乙烯基吡啶/甲基丙烯酸甲酯/甲基丙烯酸共聚物、2-甲基-5-乙烯基吡啶/丙烯酸甲酯/甲基丙烯酸共聚物、2-甲基-5-乙基吡啶/甲基丙烯酸/丙烯酸甲酯共聚物、2-乙烯基吡啶/甲基丙烯酸/丙烯腈共聚物、羧基甲基哌啶基淀粉、羧基-甲基苄基氨基纤维素、n-乙烯基甘氨酸/苯乙烯共聚物、聚葡萄胺糖、聚(乙烯醇)、顺丁烯二酸酐共聚物、聚(乙烯吡咯烷酮)、淀粉和基于淀粉的聚合物、聚(2-乙基-2-噁唑啉)、聚(乙烯亚胺)、聚氨基甲酸酯水凝胶、文莱胶、鼠李聚糖胶、聚乙酸乙烯酯、乙基纤维素、丙烯酸树脂rl、rs、ne30d和kollicoatemm30d或其组合。
在一些实施方案中,与其他上文或下文实施方案结合,所述控制释放剂是聚合物。
在一些实施方案中,与其他上文或下文实施方案结合,所述控制释放剂选自普鲁兰、糊精、钠和钙酸、聚丙烯酸、聚甲基丙烯酸、聚甲基乙烯醚共-顺丁烯二酸酐、聚乙烯吡咯烷酮、聚氧化乙烯、聚乙二醇、羟丙基纤维素、羟丙基甲基纤维素、羟基乙基纤维素、甲基丙烯酸羟基甲酯、羧基甲基纤维素钠、羧基甲基纤维素钙、甲基纤维素、麦芽糊精、三仙胶、黄蓍胶、琼脂、结冷胶、卡亚拉胶、褐藻酸、果胶、预胶凝化淀粉、聚乙烯醇、羧基甲基乙基纤维素、邻苯二甲酸乙酸纤维素、乙酸丁二酸纤维素、邻苯二甲酸甲基纤维素、邻苯二甲酸羟甲基乙基纤维素、邻苯二甲酸羟丙基甲基纤维素、乙酸丁二酸羟丙基甲基纤维素、聚乙烯醇邻苯二甲酸酯、聚丁酸乙烯邻苯二甲酸酯、聚乙烯缩醛邻苯二甲酸酯、乙酸乙烯酯/顺丁烯二酸酐共聚物、苯乙烯/顺丁烯二酸单酯共聚物、丙烯酸甲酯/甲基丙烯酸共聚物、苯乙烯/丙烯酸共聚物、丙烯酸甲酯/甲基丙烯酸/丙烯酸辛酯共聚物、甲基丙烯酸/甲基丙烯酸甲酯共聚物、苄基氨基甲基纤维素、二乙基氨基甲基纤维素、哌啶基乙基羟基乙基纤维素、乙酸二甲基氨基乙酸纤维素、乙烯基二乙胺/乙酸乙烯酯共聚物、乙烯基苄胺/乙酸乙烯酯共聚物、聚乙烯缩醛二乙基氨基乙酸酯、乙烯基哌啶基乙酰缩醛/乙酸乙烯酯共聚物、聚二乙基氨基甲基苯乙烯、甲基丙烯酸甲酯/甲基丙烯酸丁酯/甲基丙烯酸二甲基氨基乙酯和聚甲基丙烯酸二甲基氨基乙酯的共聚物、2-甲基-5乙烯基吡啶/甲基丙烯酸甲酯/甲基丙烯酸共聚物、2-甲基-5-乙烯基吡啶/丙烯酸甲酯/甲基丙烯酸共聚物、2-甲基-5-乙烯基吡啶/甲基丙烯酸/丙烯酸甲酯共聚物、2-乙烯基吡啶/甲基丙烯酸/丙烯腈共聚物、羧基甲基哌啶基淀粉、羧基-甲基苄基氨基纤维素、n-乙烯基甘氨酸/苯乙烯共聚物、聚葡萄胺糖、聚(乙烯醇)、顺丁烯二酸酐共聚物、聚(乙烯吡咯烷酮)、淀粉和基于淀粉的聚合物、聚(2-乙基-2-噁唑啉)、聚(乙烯亚胺)、聚氨基甲酸酯水凝胶、文莱胶、鼠李聚糖胶、聚乙酸乙烯酯、乙基纤维素、丙烯酸树脂rl、rs、ne30d和kollicoatemm30d或其任何组合。
ph调节剂
如本文所用,术语“ph调节剂”指的是能够调节ph至所需范围的药剂。在一些实施方案中,与其他上文或下文实施方案结合,ph调节剂为酸化剂。在一些实施方案中,与其他上文或下文实施方案结合,ph调节剂以足以降低ph的量存在。ph调节剂包括顺丁烯二酸、柠檬酸、酒石酸、双羟萘酸、反丁烯二酸、水杨酸、2,6-二氨基己酸、樟脑磺酸、甘油磷酸、2-羟基乙烷磺酸、羟乙基磺酸、丁二酸、碳酸、对甲苯磺酸、天冬氨酸、8-氯-茶碱、苯磺酸、苹果酸、乳清酸、草酸、苯甲酸、2-萘磺酸、硬脂酸、己二酸、对氨基-水杨酸、5-氨基水杨酸、抗坏血酸、硫酸、环己胺磺酸、月桂基硫酸钠、葡糖庚酸、葡糖醛酸、甘氨酸、硫酸、杏仁酸、1,5-萘二磺酸、烟碱酸、油酸、2-氧代戊二酸、5-磷酸吡哆醛、十一烷酸、对乙酰氨基苯甲酸、邻乙酰氨基-苯甲酸、间乙酰氨基苯甲酸、n-乙酰基-l-天冬氨酸、樟脑酸、去氢胆酸、丙二酸、依地酸、乙二胺四乙酸、乙基硫酸、羟基苯基苯甲酰基苯甲酸、谷氨酸、甘草酸、4-己基间苯二酚、马尿酸、对苯酚磺酸、4-羟基苯甲酸、3-羟基苯甲酸、3-羟基-2-萘甲酸、1-羟基-2萘甲酸、乳糖酸、3’-腺苷酸、5’-腺苷酸、粘液酸、半乳糖二酸、泛酸、果胶酸、聚半乳糖醛酸、5-磺基水杨酸、1,2,3,6-四氢-1,3-二甲基-2,6-二氧代嘌呤-7-丙烷磺酸、对苯二甲酸、1-羟基-2萘甲酸和其组合。在一些实施方案中,与其他上文或下文实施方案结合,所述酸性赋形剂包括例如顺丁烯二酸、柠檬酸、苹果酸、反丁烯二酸、硫酸、酒石酸、乳酸、水杨酸、天冬氨酸、氨基水杨酸、丙二酸、谷氨酸和其组合。
在一些实施方案中,与其他上文或下文实施方案结合,ph调节剂包括顺丁烯二酸、柠檬酸、酒石酸、双羟萘酸、反丁烯二酸、水杨酸、2,6-二氨基己酸、樟脑磺酸、甘油磷酸、2-羟基乙烷磺酸、羟乙基磺酸、丁二酸、碳酸、对甲苯磺酸、天冬氨酸、8-氯-茶碱、苯磺酸、苹果酸、乳清酸、草酸、苯甲酸、2-萘磺酸、硬脂酸、己二酸、对氨基-水杨酸、5-氨基水杨酸、抗坏血酸、硫酸、环己胺磺酸、月桂基硫酸钠、葡糖庚酸、葡糖醛酸、甘氨酸、硫酸、杏仁酸、1,5-萘二磺酸、烟碱酸、油酸、2-氧代戊二酸、5-磷酸吡哆醛、十一烷酸、对乙酰氨基苯甲酸、邻乙酰氨基-苯甲酸、间乙酰氨基苯甲酸、n-乙酰基-l-天冬氨酸、樟脑酸、去氢胆酸、丙二酸、依地酸、乙二胺四乙酸、乙基硫酸、羟基苯基苯甲酰基苯甲酸、谷氨酸、甘草酸、4-己基间苯二酚、马尿酸、对苯酚磺酸、4-羟基苯甲酸、3-羟基苯甲酸、3-羟基-2-萘甲酸、1-羟基-2萘甲酸、乳糖酸、3’-腺苷酸、5’-腺苷酸、粘液酸、半乳糖二酸、泛酸、果胶酸、聚半乳糖醛酸、5-磺基水杨酸、1,2,3,6-四氢-1,3-二甲基-2,6-二氧代嘌呤-7-丙烷磺酸、对苯二甲酸、1-羟基-2萘甲酸和其组合。
在一些实施方案中,与其他上文或下文实施方案结合,ph调节剂选自顺丁烯二酸、柠檬酸、苹果酸、反丁烯二酸、硫酸、酒石酸、乳酸、水杨酸、天冬氨酸、氨基水杨酸、丙二酸、谷氨酸和其任何组合。
在一些实施方案中,与其他上文或下文实施方案结合,反丁烯二酸优选作为ph调节剂,因为其与柠檬酸相比具有较少吸湿性且更可与奥美卡替莫卡必尔二盐酸盐水合物相容,引起较少或不引起活性形式转化且当在40℃/75%rh下存储6个月时片剂外观无变化,产生改进的最终产物品质。另外,测得反丁烯二酸比柠檬酸更具酸性(2倍)。因此,更有效的是使用反丁烯二酸来调节微环境ph以增强中性环境下的奥美卡替莫卡必尔释放,即,与活性物质的1:1重量比来代替2:1。反丁烯二酸还具有极低溶解速率。因而,反丁烯二酸将在片剂中待得更久且更好地维持低微环境ph,使得奥美卡替莫卡必尔在24小时内更完全释放。
填充剂
如本文所用,术语“填充剂”指的是可添加至药物组合物的组分中以增加待配置(例如,制片)材料的整体重量以便实现所需重量的一种或多种物质。填充剂包括但不限于淀粉、乳糖、甘露糖醇(如pearlitoltmsd200)、纤维素衍生物、磷酸钙、糖等。
不同等级的乳糖包括但不限于乳糖一水合物、乳糖dt(直接制片)、无水乳糖、flowlactm(可购自meggleproducts)、pharmatosetm(可购自dmv)和其他乳糖。不同等级的淀粉包括但不限于玉米淀粉、马铃薯淀粉、米淀粉、小麦淀粉、预胶凝化淀粉(可作为pcspc10购自signetchemicalcorporation)和starch1500、starch1500lm级(低水分含量级,来自colorcon)、完全预胶凝化淀粉(可作为national78-1551购自essexgrainproducts)和其他淀粉。可使用的不同纤维素化合物包括结晶纤维素和粉末状纤维素。结晶纤维素产品的实例包括但不限于ceolustmkg801、aviceltmph101、ph102、ph301、ph302及ph-f20、微晶纤维素114和微晶纤维素112。其他适用的填充剂包括但不限于羧甲纤维素、糖醇(如甘露糖醇、山梨糖醇和木糖醇)、碳酸钙、碳酸镁、磷酸氢钙和磷酸三钙。
在一些实施方案中,与其他上文或下文实施方案结合,所述填充物选自淀粉、乳糖、甘露糖醇(如pearlitoltmsd200)、纤维素衍生物、磷酸钙和糖。
在一些实施方案中,与其他上文或下文实施方案结合,所述填充物是无水乳糖或乳糖单水化物。在一些实施方案中,与其他上文或下文实施方案结合,所述填充物是乳糖dt、flowlactm或pharmatosetm。
在一些实施方案中,与其他上文或下文实施方案结合,所述填充物是玉米淀粉、马铃薯淀粉、米淀粉、小麦淀粉、预胶凝化淀粉(starch1500、starch1500lm级(低水分含量级))、完全预胶凝化淀粉。
在一些实施方案中,与其他上文或下文实施方案结合,所述填充物是微晶纤维素如ceolustmkg801、aviceltmph101、ph102、ph301、ph302和ph-f20、微晶纤维素114或微晶纤维素112。
在一些实施方案中,与其他上文或下文实施方案结合,所述填充物是羧甲纤维素、甘露糖醇、山梨糖醇、木糖醇、碳酸钙、碳酸镁、磷酸氢钙和磷酸三钙。
润滑剂
如本文所用,术语“润滑剂”指的是可添加至本发明组合物的组分中以降低固体制剂对用于生产单位剂型的设备的粘着的一种或多种物质。润滑剂包括硬脂酸、氢化植物油、氢化大豆油和氢化大豆油与蓖麻蜡、硬脂醇、白氨酸、聚乙二醇、硬脂酸镁、单硬脂酸甘油酯、硬脂酸、山嵛酸甘油酯、聚乙二醇、环氧乙烷聚合物、月桂基硫酸钠、月桂基硫酸镁、油酸钠、硬脂酰反丁烯二酸钠、dl-白氨酸、胶态二氧化硅和其混合物。
在一些实施方案中,与其他上文或下文实施方案结合,润滑剂包括硬脂酸、氢化植物油、氢化大豆油和氢化大豆油与蓖麻蜡、硬脂醇、白氨酸、聚乙二醇、硬脂酸镁、单硬脂酸甘油酯、硬脂酸、山嵛酸甘油酯、聚乙二醇、环氧乙烷聚合物、月桂基硫酸钠、月桂基硫酸镁、油酸钠、硬脂酰反丁烯二酸钠、dl-白氨酸、胶态二氧化硅和其任何混合物。
制造方法
也提供一种用于制备如本文所述的药物制剂的方法,其包括:
共混包含奥美卡替莫卡必尔或其药学上可接受的盐、药学上可接受的水合物或药学上可接受的盐的药学上可接受的水合物、控制释放剂、ph调节剂以及填充剂的混合物;
使用润滑剂润滑所述共混的混合物;
使润滑的共混物形成造粒;
使用润滑剂润滑所得造粒物;以及
将润滑的造粒物压缩成所需的形式。
还提供一种用于制备如本文所述的药物制剂的方法,其包括:
提供包含奥美卡替莫卡必尔或其药学上可接受的盐、药学上可接受的水合物或药学上可接受的盐的药学上可接受的水合物、控制释放剂、ph调节剂以及填充剂的共混的混合物;
将共混的混合物造粒;以及
将润滑的造粒物压缩成所需的形式。
还提供一种用于制备如本文所述的药物制剂的方法,其包括:
将奥美卡替莫卡必尔或其药学上可接受的盐、药学上可接受的水合物或药学上可接受的盐的药学上可接受的水合物、控制释放剂、ph调节剂、填充剂以及润滑剂的造粒物压缩成所需的形式。
在一些实施方案中,与其他上文或下文实施方案结合,改性的释放基质片剂使用干式造粒来制造。所述干式造粒方法可帮助避免改性的释放基质片剂中的活性形式转化。此外,干式造粒方法避免在高剪切湿式造粒方法中观测到的问题。
还提供如本文所述的通过任何方法制备的药物制剂。
稳定性
使用强制降解条件(例如40℃和75%相对湿度)来评估药物成分或组合物的长期储存稳定性。一般而言,稳定组合物为在经受强制降解条件后包含药学上活性成分的组合物,所述药学上活性成分的量相对于最初存在于特定组合物中的量为例如95%。稳定性可使用强制降解或其他方法持续1周、1个月、2个月、3个月、4个月、5个月、6个月、9个月、12个月、15个月、18个月、24个月、30个月、36个月、更长时期来测定。
药物技术中已知评估药物组合物稳定性的分析法,如本发明中所述的那些分析法。例如,可通过使用标准分析技术来测定存在于给定组合物中的活性药物成分的百分比,以及杂质的存在和百分比。
公开的处理方法/制剂的用途
也提供一种用于使用这类药物制剂治疗心脏衰竭的方法,所述心脏衰竭包括但不限于:急性(或失代偿性)充血心脏衰竭以及慢性充血心脏衰竭;特别是与收缩心脏功能障碍相关的疾病。
实例
制造奥美卡替莫卡必尔二盐酸盐水合物
奥美卡替莫卡必尔的合成途径
合成apism哌嗪硝基-hcl
一般方法
试剂和溶剂自商业来源以原样使用。在400mhz光谱仪上记录1hnmr光谱。使用溶剂共振作为内标(cdcl3,dmso-d6),以ppm形式报告从四甲基硅烷的化学位移。数据报告如下:化学位移、多重性(s=单峰,d=双峰,t=三峰,q=四峰,br=宽峰,m=多重峰)、偶合常数(hz)和积分。在100mhz光谱仪上使用完全质子去耦记录13cnmr光谱。使用溶剂作为内部参考(cdcl3,dmso-d6),以ppm形式报告从四甲基硅烷的化学位移。关于起始2-氟代-3-硝基甲苯进行所有溶剂馈料。
x射线粉末衍射数据(xrpd)是使用配备有即时多带(rtms)检测器的panalyticalx’pertpro衍射仪(panalytical,almelo,thenetherlands)来获得。所用辐射为
或者,xrpd数据是使用配备有rtms检测器的panalyticalx’pertpro衍射仪(panalytical,almelo,thenetherlands)来获得。所用辐射为
或者,xrpd数据是使用配备有rtms检测器的panalyticalx’pertpro衍射仪(panalytical,almelo,thenetherlands)来获得。所用辐射为
或者,xrpd数据是使用配备有rtms检测器的panalyticalx’pertpro衍射仪(panalytical,almelo,thenetherlands)来获得。所用辐射为
或者,xrpd数据是使用配备有电动xyz样品台和gadds面积检测器的brukerd8discoverx射线衍射系统(bruker,billerica,ma)来获得。所用辐射为
差示扫描热量测定(dsc)数据是使用标准dsc模式(dscq200,tainstruments,newcastle,de)来收集。在40℃至300℃的温度范围内采用10℃/min的加热速率。在氮气下进行分析并且样品被装载于标准密封铝盘中。铟用作校准标准。
或者,dsc数据是使用温度调变dsc模式(dscq200,tainstruments,newcastle,de)来收集。在20℃下样品平衡5分钟之后,在20℃至200℃的温度范围内,以+/-0.75℃/min调变采用3℃/min的加热速率。在氮气下进行分析并且样品被装载于标准未卷曲铝盘中。铟用作校准标准。
fn-溴化物
在配备有回流冷凝器和洗涤器并且馈有5nnaoh溶液的60l反应器(不含暴露的不锈钢、
分析产率为2.5%fn-甲苯、62.3%fn-溴化物和30.0%二-溴化物。甲苯溶液不含过氧化苯甲酰、丁二酰亚胺或α-溴乙酸,并且通过kf滴定测得的水含量为1030ppm(这个溶液可在氮气下在室温下保持>12h,而无分析产率的任何变化)。
在室温下,向这个溶液中添加二异丙基乙胺(880.0g,6.63mol,0.53当量),随后添加甲醇(460ml,11.28mol,0.88当量)且加热至40℃。制备亚磷酸二乙酯(820.0g,5.63mol,0.46当量)在甲醇(460ml,11.28mol,0.88当量)中的溶液且在40℃下通过加料漏斗经1小时时期以使得批料温度在40±5℃内的速率添加至反应混合物中。从开始添加时起,在40℃下搅拌内含物3h时期且冷却至室温,并在氮气氛围下保持12小时。反应混合物的分析产率为2.5%fn-甲苯、92.0%fn-溴化物和0.2%二-溴化物。这个溶液原样用于烷基化步骤。
表征最终产物混合物的组分(针对纯化合物进行收集)。
2-氟代-3-硝基甲苯(fn-甲苯):1hnmr(400mhz,氯仿-d)δppm2.37(s,1h),7.13-7.20(m,1h),7.45-7.51(m,1h),7.79-7.85(m,1h)。13cnmr(100mhz,氯仿-d)δppm14.3(d,j=5hz),123.3(d,j=3hz),123.6(d,j=5hz),128.2(d,j=16hz),136.7(d,j=5hz),137.5(宽峰),153.7(d,j=261hz);1-(溴甲基)-2-氟代-3-硝基苯(fn-溴化物):1hnmr(400mhz,氯仿-d)δppm4.56(s,1h),7.28-7.34(m,1h),7.69-7.76(m,1h),7.98-8.05(m,1h)。13cnmr(100mhz,氯仿-d)δppm23.6(d,j=5hz),124.5(d,j=5hz),126.1(d,j=3hz),128.5(d,j=14hz),136.5(d,j=4hz),137.7(宽峰),153.3(d,j=265hz)。dsc:在53.59℃下单一熔体。精确质量[c7h5brfno2+h]+:计算值=233.9566,测量值=233.9561;1-(二溴甲基)-2-氟代-3-硝基苯(二溴化物):1hnmr(400mhz,氯仿-d)δppm6.97(s,1h),7.39-7.45(m,1h),8.03-8.10(m,1h),8.16-8.21(m,1h)。13cnmr(100mhz氯仿-d)δppm29.2(d,j=7hz),124.9(d,j=5hz),127.1(d,j=2hz),132.1(d,j=11hz),137.2(d,j=2hz),135.7(宽峰),149.8(d,j=266hz)。dsc:在49.03℃下单一熔体。精确质量[c7h4br2fno2+h]+:计算值=311.8671,测量值=311.8666。
哌嗪硝基-hcl:
在22℃下,在氮气氛围下向60l反应器中fn-溴化物(从前一步骤制备)的机械搅拌甲苯溶液(9体积)中馈入二异丙基乙胺(1.90kg,14.69mol,1.14当量)。以允许批料温度停留在30.0℃下的速率向这个混合物中添加哌嗪甲酸甲酯(哌嗪甲酸酯)(2.03kg,14.05mol,1.09当量)在甲苯(1.0l,0.5体积)中的溶液(放热。在添加期间,夹套温度被调整至5℃以便维持批料温度低于30℃。混合物在22℃下搅拌3小时并且等分试样的分析证实烷基化反应完全(<1.0lcapfn-溴化物,hplc254nm)。反应混合物用nh4cl水溶液(20wt%,10.0l,5体积;从2.0kgnh4cl和10.0ldi水制备)处理,搅拌两相混合物(30min)并且分离各层。有机层依序用nahco3水溶液(9wt%,10.0l,5体积;从0.90kgnahco3和10.0ldi水制备)洗涤。有机层通过5μm
在氮气氛围下,在22℃下,向60l反应器中的哌嗪硝基游离碱(如上所述而制备)的机械搅拌甲苯溶液中馈入ipa(19.4l,9.7体积)和di水(1.0l,0.5体积)。混合物加热至55℃且馈入20%1.4当量浓hcl(在使用之前加以滴定且基于效价值馈入;276.0ml,3.21mol)。内含物搅拌15min且以ipa(400ml,0.2体积)中的浆液形式馈入哌嗪硝基-hcl晶种(130.0g,0.39mol,0.03当量)。混合物搅拌30min并且经4小时时期添加剩余浓hcl(馈料的80%,1.10l,12.82mol)。混合物在55℃下搅拌1h,以线性方式经1.5小时冷却至20℃,并且在这个温度下搅拌12小时。测量哌嗪硝基-hcl的上清液浓度(2.8mg/g)。混合物提供配备有5μm
4-(2-氟代-3-硝基苄基)哌嗪-1-甲酸甲酯盐酸盐(哌嗪硝基-hcl):1hnmr(300mhz,dmso-d)δppm3.25(br.s,3h),3.52-3.66(m,8h),4.47(s,2h),7.44-7.63(t,1h,j=8hz),7.98-8.15(m,1h),8.17-8.34(m,1h)。13cnmr(75mhz,dmso-d)δppm50.3,51.4,52.8,119.6(d,j=14hz),125.1(d,j=5hz),127.9,137.4(d,j=8hz),139.8(d,j=3hz),152.2,154.7,155.7。dsc:在248.4℃下开始熔化。精确质量[c13h16fn3o4+h]+:计算值=298.1203,测量值=298.1198。
哌嗪硝基游离碱:
在周围温度下,在氮气氛围下,在配备有回流冷凝器的60l反应器中机械搅拌哌嗪硝基-hcl(2.0kg,5.99mol,1.0当量)和乙酸异丙酯(6.0l,3.0体积)的混合物。添加在另一容器中制备的碳酸氢钠(629g,7.49mol,1.25当量)和水(7.5l,3.75体积)的溶液。搅拌两相混合物(15min),并且分离各层。上层有机层(含有产物)转移至另一容器中,而反应器用水和异丙醇冲洗。有机层接着通过串联5μm
合成apism氨基甲酸苯酯-hcl
在设定于20℃下,氮气氛围下,且通过洗涤器排气的60l玻璃内衬夹套反应器(含有5nnaoh)中馈入2.5kg氨基吡啶(1.0当量,23.1莫耳),接着馈入25l(19.6kg,10vol)乙腈。在开始搅拌且氨基吡啶(吸热)溶解之后,在容器中馈入12.5ln-甲基-2-吡咯烷酮(12.8kg,5vol)。在加料漏斗中馈入1.8l(0.6当量,13.9mol)氯甲酸苯酯,接着经68分钟添加至氨基吡啶溶液中,保持内部温度≤30℃。反应在20±5℃的内部温度下搅拌>30分钟。在容器中接着馈入61±1g呈200ml乙腈中的浆液形式的晶种且老化≥30min。在加料漏斗中馈入1.25l(0.45当量,9.7mol)氯甲酸苯酯,接着经53分钟添加至反应悬浮液中,同时再次保持温度≤30℃。反应器的内含物在20±5℃下老化≥30小时。在分析上清液(对于产物和起始材料两者,≤15mg/g)之后,使用配备有12μmteflon布的aurora过滤器过滤固体。母液转递至第2个60l玻璃内衬夹套反应器中。反应器和滤饼用1x10l5:10nmp/acn和1x10lacn冲洗。洗涤液也转递至第二反应器中。滤饼在真空下使用氮气放气干燥≥24小时以提供5.65kg(90.2%产率)呈灰白色固体状的产物氨基甲酸苯酯-hcl,98.8wt%,99.2%lcap纯度。
(6-甲基吡啶-3-基)氨基甲酸苯酯盐酸盐(氨基甲酸苯酯-hcl)1hnmr(400mhz,dmso-d6)δppm11.24(s,1h),8.81(s,1h),8.41(d,1h,j=8.8hz),7.85(d,1h,j=8.8hz),7.48-7.44(m,2h),7.32-7.26(m,3h),2.69(s,3h);13cnmr(100mhz,dmso-d6)δppm151.66,150.01,147.51,136.14,133.79,129.99,129.49,127.75,125.87,121.70,18.55;hr-ms:c13h12n2o2计算值:228.0899,m+h+=229.0972;观测质量:229.0961
gmp步骤
4-(3-氨基-2-氟苄基)哌嗪-1-甲酸甲酯(哌嗪苯胺)
向100-l夹套玻璃内衬反应器中添加4-(2-氟代-3-硝基苄基)哌嗪-1-甲酸甲酯盐酸盐(2.00kg,1.00当量)和乙酸异丙酯(6.00l,相对于起始材料为3.00vol)。在氮气吹扫下搅拌所得浆液。经45±30min向混合物中逐滴添加:7.7%w/w碳酸氢钠水溶液(629g,1.25当量碳酸氢钠溶解于7.50l水中),同时通过夹套控制维持20±5℃之内部温度(注释:添加为吸热的,并且可析出多达1当量二氧化碳气体)。混合物搅拌≥15min,得到澄清两相混合物。停止搅拌且使各层沈降。
底(水)层排水且通过ph纸分析以确保层的ph>6。上(有机)层的定量hplc分析揭露4-(2-氟代-3-硝基苄基)哌嗪-1-甲酸甲酯游离碱(1.73-1.78kg)的97-100%分析产率。上(有机)层通过串联过滤器转移至20-l
在反应结束时,用氮气净化氢化器两次(加压至60±10psig,接着排气至大气压)。粗反应混合物依序通过5μm过滤器和0.45μm过滤器过滤至40-l玻璃内衬反应器中。氢化器和管线用额外乙酸异丙酯等分试样(2.00l)洗涤。粗反应混合物的定量hplc分析揭露95-100%分析产率(1.52-1.60kg苯胺产物)。反应混合物在减压(通常为250-300毫巴)下在50±5℃的批料温度下蒸馏,直到总反应体积为约8.00l(4.00vol)。批料通过添加庚烷以控制总批料体积而在50±5℃、250-300毫巴下经受恒定体积蒸馏。在添加约8.00l(4.00vol)庚烷的后,gc分析指示溶剂组成为约50%乙酸异丙酯、50%庚烷。打破真空且维持内部批料温度在50±5℃下。向反应混合物中添加晶种浆液(20.0g产物4-(3-氨基-2-氟苄基)哌嗪-1-甲酸甲酯,在80ml庚烷和20ml乙酸异丙酯的溶剂混合物中)。使所得浆液在50±5℃下搅拌2±1小时,接着经2.5±1.0h冷却至20±5℃。经2小时逐滴添加额外庚烷(24.0l,12.0vol),并且使批料在20±5℃下搅拌≥1小时(通常过夜)。这个过滤的上清液的定量hplc分析揭露溶液中<5mg/ml产物,产物晶体为50-400μm双折射棒。反应浆液在20℃下过滤至滤布上,并且用庚烷(6.00l,2.00vol)对滤饼进行置换洗涤。滤饼在氮气吹扫下在周围温度下在过滤器上干燥>4小时,直到通过lod分析确认样品干燥(指示<1.0wt%损失)。产物4-(3-氨基-2-氟苄基)哌嗪-1-甲酸甲酯(1.56kg)分离成浅黄色粉末状,86%产率,通过hplc测得99.8wt%,100.0lcap210。[合并的滤液和洗涤液的分析揭露母液中失去108g(7.0%)产物。剩余质量余额包含反应器中的产物滞留量(结垢)。]1hnmr(dmso-d6,400mhz)δ:6.81(dd,j=7.53,7.82hz,1h),6.67(m,1h),6.49(m,1h),5.04(s,2h),3.58(s,3h),3.45(m,2h),3.34(m,4h),2.33(m,4h)。19fnmr(d6-dmso,376mhz)δ:-140.2。13cnmr(d6-dmso,125mhz)δ:155.0,150.5,148.2,136.2(m),123.7(m),117.6,115.1,73.7,54.9(m),52.1(m),43.4。mp=89.2℃。
奥美卡替莫卡必尔二盐酸盐水合物程序
向15l玻璃内衬反应器中馈入4-(3-氨基-2-氟苄基)哌嗪-1-甲酸甲酯(1,202g,4.50mol)、(6-甲基吡啶-3-基)氨基甲酸苯酯盐酸盐(1,444g,5.40mol)和四氢呋喃(4.81l)。所得浆液在氮气吹扫下搅拌且接着n,n-二异丙基乙胺(1,019l,5.85mol)馈入至浆液中,得到褐色溶液。溶液温度增至65℃且搅拌22h,直到通过hplc分析测得保留<1%auc哌嗪苯胺。
批料冷却至50℃且在减压下蒸馏,同时通过调整真空压力维持容器的内部温度低于50℃。在残余真空下以一定速率添加2-丙醇以维持15l反应器中的恒定体积。需要总计10.5kg2-丙醇以实现通过gc测得之<5%thf。水(2.77kg)接着馈入至反应器中,接着以一定速率添加6nhcl(1.98kg)以维持内部温度低于60℃。在氮气吹扫下使反应器达到周围压力。溶液接着加热至60℃,并且经由串联过滤器转移至60l玻璃内衬反应器中。15l反应器接着用1:1水/2-丙醇(1.2l)冲洗,通过串联过滤器送至60l反应器中。
60l反应器被调整至45℃且晶种(114g,0.23mol)在2-丙醇(0.35l)中的浆液添加至反应器中,形成浆液。批料在45℃下老化1h,接着通过串联过滤器经2h添加2-丙醇(3.97kg)。批料经1h加热至55℃且保持0.25h,接着经1h冷却回45℃且在45℃下保持过夜。2-丙醇(11.71kg)接着通过串联过滤器经3h添加至批料中。批料老化1h且接着经2h冷却至20℃,并且在20℃下保持0.5h。批料接着经由贴附有1-介质和2-在56hz下操作2.15h的精细转子-定子的湿式研磨机再循环,直到通过显微法不能再观测到粒径减小。
批料接着通过配备有12um滤布的20”
dsc:t开始=61.7℃,t最大=95.0℃;tga=2.2%,降解开始=222℃;1hhmr(d2o,500mhz)8.87(s,1h),8.18(d,j=8.9hz,1h),7.83(t,j=7.5hz,1h),7.71(d,j=8.8hz,1h),7.35–7.29(m,2h),4.48(s,2h),4.24(brs,2h),3.73(s,3h),3.31(brs,6h),2.68(s,3h);13chmr(d2o,150mhz)156.8,154.2,153.9(j=249hz),147.8,136.3,136.1,130.1,129.4,128.0,127.2,125.5(j=11.8hz),125.1(j=4.2hz),116.1(j=13.5hz),53.54,53.52,53.49,50.9,40.5,18.2。
比较实施例1:立即释放制剂
表1
包含以上组分的立即释放制剂是根据图1中所概述的方法来制备。
实施例1:原型改性释放制剂
奥美卡替莫卡必尔原型改性释放(“mr”)基质片剂制剂是由奥美卡替莫卡必尔无水物游离碱(活性)、methoceltmk100mcr(控制释放剂)、柠檬酸一水合物(ph调节剂)、微晶纤维素和乳糖一水合物(填充剂)、methoceltme5lv(粘合剂)以及硬脂酸镁(润滑剂)组成。表1展示原型制剂组合物。原型mr基质片剂通过常规高剪切造粒方法来制造。这种方法包括通过#20筛目us标准筛网来筛选奥美卡替莫卡必尔无水物、乳糖一水合物ffl316、微晶纤维素、
以下个案研究例示了奥美卡替莫卡必尔无水物25mg原型mr基质片剂的制造方法的实施方案。目标批料大小为60kg。针对批料计量的原材料为4.30kg奥美卡替莫卡必尔无水物(约14.7%过量以补偿粉碎损失)、10.1kg微晶纤维素(
粘合剂溶液制备:填充19.4kg的纯化水至19加仑的便携式混合锅中,并且接着缓慢并稳定地添加0.6kg的methoceltme5lv。
装载原材料至diosnap-300高剪切造粒机中:手动装载大部分筛选的乳糖一水合物和微晶纤维素至造粒机碗中。手动装载柠檬酸一水合物至碗中。手动装载研磨的奥美卡替莫卡必尔无水物至碗中。手动装载筛选的methoceltmk100mcr至碗中。
转移粘合剂溶液:转移粘合剂溶液至溶液槽中。
湿式造粒:转移6.60kg的粘合剂溶液至造粒机碗中。
流化床干燥:干式造粒。
干式研磨:手动加入干燥颗粒且开始研磨。
润滑:装载大约一半研磨的颗粒至v共混器中且接着在其中添加硬脂酸镁,在其中添加剩余一半研磨的颗粒。
压缩:将最终共混物手动加入至配备有7/16”圆形、标准杯状、凹入、平面工具的旋转压锭机的加料斗中。目标片剂重量是400mg(在370mg-430mg的范围内),目标硬度是12kp(具有10kp-14kp的范围)。
原型mr基质片剂制剂组合物
基质改性释放片剂:一般方法
本文描述通过干式造粒工艺制造改性释放(“mr”)基质片剂的工艺。筛选奥美卡替莫卡必尔二盐酸盐水合物、微晶纤维素、乳糖一水合物、methoceltmk100mcr/methoceltmk100lvcr和反丁烯二酸,并且接着将筛选的材料馈入至滚动式共混机中且在其中以预定速度共混特定时间(共混机大小、共混速度和共混时间为标度依赖性参数)。共混的材料在同一共混机中使用预筛选的硬脂酸镁润滑。润滑的共混物接着进行辊压实且研磨。所得颗粒在滚动式共混机中使用预筛选的硬脂酸镁润滑。润滑的颗粒使用旋转压锭机以10kp的目标片剂硬度压缩成改性释放基质片剂。
实施例2:奥美卡替莫卡必尔二盐酸盐水合物25mg缓慢释放mr基质片剂(mtx-f1)。
目标批料大小为5kg,针对批料计量的原材料为306.50g奥美卡替莫卡必尔二盐酸盐水合物、1840.50g微晶纤维素
粉末筛选:步骤1.通过20目us标准筛网筛选1840.5g微晶纤维素
粉末共混:步骤2.将来自步骤1的筛选的共混物馈入至20lbohle共混机中且在20rpm的速度下共混30分钟。
粉末润滑:步骤3.通过60目us标准筛网筛选全部量的粒内硬脂酸镁,并且称出所需量的过筛硬脂酸镁25.0g至适当容器中。步骤4.在同一容器中手动预混合所需量的过筛硬脂酸镁与来自步骤2的约1x至3x粉末共混物,持续约60秒。步骤5.将来自步骤4的预混合共混物馈入至步骤2中的粉末共混物中。步骤6.在20rpm的共混速度下共混来自步骤2的粉末共混物,持续4分钟。步骤7.排放润滑的粉末共混物至适当容器中。
干式造粒:步骤8.将来自步骤7的润滑粉末共混物馈入至gerteis辊压实机加料斗中且使用以下工艺参数开始干式颗粒制造。辊表面:knurl;搅拌器速度:15rpm;轧制力:7.0kn/cm;轧制速度:2rpm;辊隙:2.5mm;间隙控制:开;筛网大小:1mm;造粒机与筛网之间的间隔:2.0mm;造粒机速度:80rpm;以及造粒机旋转角:200/230°。步骤9.排放颗粒至适当容器中且称重净重量,其为4844g。
颗粒润滑:步骤10.计算颗粒共混物所需的硬脂酸镁的所需量,其为24.34g。步骤11.通过60目us标准筛网筛选全部量的粒内硬脂酸镁,并且称出所需量的步骤10中筛选的硬脂酸镁。步骤12.将来自步骤9的颗粒馈入至20升bohle共混机中。步骤13.在适当容器中手动预混合来自步骤11的筛选的粒外硬脂酸镁与1x至3x来自步骤12的颗粒,持续约60秒。步骤14.将来自步骤13的预混合共混物馈入至步骤12中的共混机中。步骤15.在20rpm的共混速度下共混来自步骤12的颗粒共混物,持续5分钟。步骤16.排放来自步骤15的颗粒共混物至适当容器中。
片剂压缩:步骤17.来自步骤16的最终颗粒共混物手动馈入至配备有7/16”圆形、标准杯状、凹入、平面工具的旋转压锭机korschxl100的加料斗中。步骤18.压缩以25rpm的速度开始至目标片剂重量和硬度的刻度盘中。目标片剂重量为500mg(在475-525mg范围内),目标硬度为10kp(具有6-14kp范围)。所制造片剂的总数为9,115个。
表2.根据本发明的奥美卡替莫卡必尔二盐酸盐水合物25mg缓慢释放mr基质片剂mtx-f1的组合物
包含以上组分的基质改性释放片剂是根据图2中所概述的方法来制备。注释:在一些实施方案中,浓度范围为15%-80%(针对methoceltmk100mcr)、0%-70%(针对微晶纤维素
实施例3
表3.根据本发明的奥美卡替莫卡必尔二盐酸盐水合物25mg快速释放mr基质片剂mtx-f2的组合物
包含以上组分的基质改性释放片剂是根据图3中所概述的方法来制备。注释:在一些实施方案中,浓度范围为0%-15%(针对methoceltmk100mcr)、15%-50%(针对methoceltmk100lv)、0%-75%(针对微晶纤维素
实施例4
表4.根据本发明的奥美卡替莫卡必尔二盐酸盐水合物75mg缓慢释放mr基质片剂mtx-f3的组合物
包含以上组分的基质改性释放片剂是根据图3中所概述的方法来制备。注释:在一些实施方案中,浓度范围为15%-80%(针对methoceltmk100mcr)、0%-65%(针对微晶纤维素
实施例5
表5.根据本发明的奥美卡替莫卡必尔二盐酸盐水合物75mg快速释放mr基质片剂mtx-f4的组合物
包含以上组分的基质改性释放片剂是根据图3中所概述的方法来制备。注释:在一些实施方案中,浓度范围为0%-15%(针对methoceltmk100mcr)、15%-50%(针对methoceltmk100lv)、0%-65%(针对微晶纤维素
ph依赖性释放分布
制备奥美卡替莫卡必尔半水合物(游离碱)和二盐酸盐水合物(形式a)的制剂,其具有以下组分,所有组分均以w/w%报告:
游离碱(75mg基质片剂)活性颗粒:15.37%游离碱;30%羟丙基甲基纤维素hpmck100mpremcr;10%柠檬酸一水合物;11.88%微晶纤维素avicelph101;6.75%乳糖一水合物fastflo316;12.5%纯化水;且柠檬酸颗粒:20%柠檬酸一水合物;5%微晶纤维素avicelph101;以及1%硬脂酸镁,非牛。
形式a(75mg基质片剂)颗粒内:18.37%形式a;30%羟丙基甲基纤维素hpmck100mpremcr;0.50%硬脂酸镁;且颗粒外:16.88%微晶纤维素avicelph101;18.37%无水柠檬酸;以及0.5%硬脂酸镁,非牛。
在ph2和ph6.8下测试制剂且测量随时间释放的药物的量。这种药物释放分布的结果展示于图6中。
前述内容仅仅说明本发明且不欲将本发明局限于所公开的化合物。本领域技术人员显而易知的变化和改变在随附权利要求书所界定的本发明范围和性质内。
- 还没有人留言评论。精彩留言会获得点赞!
- 具有吡唑母核的小分子杂环化合物及其用于制备抗菌、抗肿瘤药物的应用的制造方法与工艺
- 2‑苯基‑4‑对羟基苯基噻唑的合成方法与流程
- 一种溶剂参与反应合成4‑乙酰基‑1,2,3‑三唑化合物的方法与流程
- 一类具有抗癌活性的咪唑类化合物及其衍生物的制造方法与工艺
- 一种高酸度离子液体催化合成2‑芳基‑2,3‑二氢‑4(1H)‑喹啉酮衍生物的方法与流程
- 一种提高有机废水中COD去除效率、生化性的方法与流程
- 一种以亚氧化钛为催化剂的催化臭氧化水处理方法与流程
- 一种基于UIO‑66与铜纳米线原位共组装合成吸附‑光催化复合材料的制造方法与工艺
- 4N杂环化合物的衍生物在制备用于治疗自身免疫疾病的药物中的应用的制造方法与工艺
- 苯并氮杂*磺酰胺化合物的制造方法与工艺
- 含氮的稠环化合物以及使用其的有机发光器件的制造方法与工艺
- (杂)芳基1,3‑偶极化合物与(杂)环炔的环加成方法与制造工艺
- 一种具抗肿瘤活性的岩白菜素氮杂肉桂酸酯类化合物及其合成方法与制造工艺
- 一种含有氮杂环的化合物及其有机电致发光器件的制造方法与工艺
- 靛红杂合喹唑啉类化合物合成的靛红衍生物及其在制备抗肿瘤药物中的应用的制造方法与工艺
- 氮杂环类化合物及其应用的制造方法与工艺
- 含唑杂环的嘧啶类化合物,组合物及其用途的制造方法与工艺
- 一种具有抗肿瘤活性的高乌甲素氮杂肉桂酸杂合体及其合成方法与制造工艺
- 一种氮杂芳烃苄位与缺电子芳环偶联的方法与制造工艺
- 一种含氮杂环衍生物及使用该含氮杂环衍生物的有机发光器件的制造方法与工艺