一种依鲁替尼的新晶型及其制备方法与流程
本发明涉及化学制药,具体涉及药物制备,尤其涉及依鲁替尼,1-((r)-3-(4-氨基-3-(4-苯氧基苯基)-1h-吡唑并[3,4-d]嘧啶-1-基)哌啶-1-基)丙-2-烯-1-酮的新晶型及其制备方法。
背景技术:
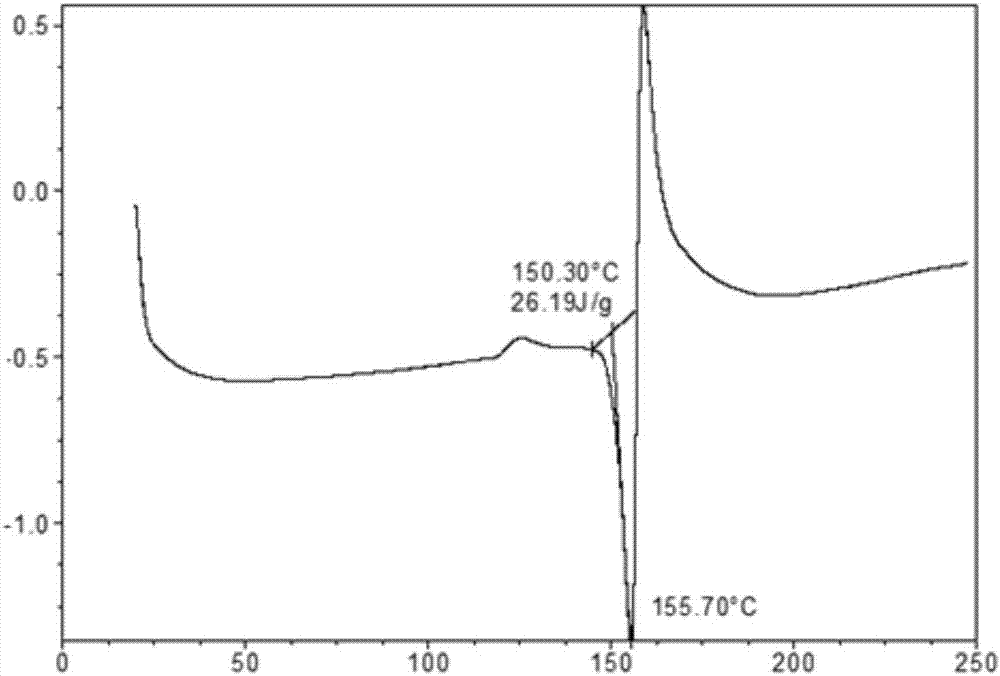
:依鲁替尼(ibrutinib)是一种选择性地抑制布鲁顿酪氨酸激酶(btk)的靶向性抑制剂,由美国pharmacyclics公司开发。其化学名为1-((r)-3-(4-氨基-3-(4-苯氧基苯基)-1h-吡唑并[3,4-d]嘧啶-1-基)哌啶-1-基)丙-2-烯-1-酮。结构如式i所示:布鲁顿酪氨酸激酶(btk)是一种非受体酪氨酸激酶,是b-淋巴细胞形成、分化、信息传递和生存所必需的重要介质。抑制布鲁顿酪氨酸激酶(btk)的活性,可以有效抑制肿瘤细胞的增殖和存活。2013年11月13日美国食品药品管理局已批准依鲁替尼可用于套细胞淋巴瘤。2014年和2015年美国食品药品管理局又批准增加了依鲁替尼新的适应症:慢性淋巴性白血病及华氏巨球蛋白血症。药物分子可能存在不同的晶型,据报道依鲁替尼有多种晶型。pharmacyclics公司的中国专利cn104736178a中公开了依鲁替尼的六种晶型:无水合物的晶型a、晶型b、晶型c以及溶剂合物晶型d、晶型e、晶型f。晶型a的xrpd谱图在2θ值为5.7±0.1°、13.6±0.1°、16.1±0.1°、18.9±0.1°、21.3±0.1°和21.6±0.1°处具有特征峰;晶型b的xrpd谱图在2θ值为5.2±0.1°、10.2±0.1°、16.5±0.1°、18.5±0.1°和20.8±0.1°处具有特征峰;晶型c的xrpd谱图在2θ值为7.0±0.1°、14.0±0.1°、15.7±0.1°、18.2±0.1°、19.1±0.1°、19.5±0.1°、20.3±0.1°、22.1±0.1°和22.9±0.1°处具有特征峰;晶型d为甲基异丁基酮溶剂合物;晶型e为甲苯溶剂合物;晶型f为甲醇溶剂合物。中国专利cn104327085a中公开了一种新的依鲁替尼无水合物晶型a,其具有较好的溶解性和稳定性等性质。这种晶型的特征在于,其xrpd谱图在2θ值为5.2°±0.2°、17.6°±0.2°、22.1°±0.2°、19.3°±0.2°、22.4°±0.2°、20.8°±0.2°处具有特征峰。中国专利cn103923084a公开了几种依鲁替尼晶型:无水合物晶型ii,水合物晶型iii,四氢呋喃溶剂合物晶型iv以及三氯甲烷溶剂合物晶型v、晶vi、晶型vii。然而,药物分子不同晶型具有不同的物理、化学性质,其溶解性、稳定性等可能有显著的差异,影响到药物的生物利用度、疗效和安全性等,因此,寻找理化性质更加优异的晶型,提升药物的有效性、安全性和质量可控性具有重大的意义。技术实现要素:本发明所要解决的技术问题是研究设计生物利用度、疗效和安全性更好的依鲁替尼的新晶型。本发明提供了一种依鲁替尼的新晶型,命名为晶型1。本发明提供的依鲁替尼新晶型,其x射线粉末衍射图2θ值在5.8°±0.2°、10.8°±0.2°、19.2°±0.2°、21.6°±0.2°处具有特征峰。本发明提供的依鲁替尼新晶型,其具有基本如图1所示的x射线粉末衍射(xrpd)图。本发明提供的依鲁替尼新晶型,其在加热至150.3℃附近开始出现吸热峰。本发明提供的依鲁替尼新晶型,其具有基本如图2所示的差示扫描量热(dsc)分析图。本发明提供的依鲁替尼新晶型,其具有基本如图3所示的热重分析(tga)图。本发明提供的依鲁替尼新晶型,其为无水物。本发明提供的依鲁替尼新晶型,其具有基本如图4所示的动态水分吸附(dvs)图。本发明提供的依鲁替尼新晶型,其为非吸湿性的。本发明的另一目的是提供了依鲁替尼晶型1的制备方法:方法a、方法b或方法c。本发明提供一种依鲁替尼晶型1的制备方法(方法a),包括下列步骤:a1)将依鲁替尼粉末加入甲醇中,加热至温度1,搅拌使溶液澄清;a2)降温至室温,加入水,搅拌;a3)降温至温度2,搅拌,析晶,过滤收集固体,得到晶型1。其中:步骤a1)中,所述依鲁替尼采用与cn101610676a实施例1b相同的方法制备;步骤a1)中,所述依鲁替尼与甲醇的用量比为50~250:1(mg/ml),优选为50~150:1(mg/ml),更优选为80~150:1(mg/ml);步骤a1)中,所述温度1为40℃~65℃,优选为40~60℃,更优选为50℃;步骤a1)中,所述搅拌的时间为1~12小时,优选为1~5小时,更优选为1~3小时;步骤a2)中,所述室温为20℃~25℃,优选为25℃;步骤a2)中,所述降温的速度在0.5~2小时内使温度降至所述室温,优选在1~2小时内将温度降至所述室温;步骤a2)中,所述水与步骤a1)中所述甲醇的体积比为1:1~1:10,优选体积比为1:1~1:6,更优选体积比为1:1~1:4;步骤a2)中,所述搅拌的时间为1小时~12小时,优选为1小时~5小时,更优选为1小时~3小时;步骤a3)中,所述温度2为0℃~15℃,优选为5℃~15℃,更优选为5℃~10℃。步骤a3)中,所述降温的速度为每小时降温10℃~30℃,更优选降温速度为每小时降温20℃;步骤a3)中,所述搅拌的时间为0.5小时~72小时,优选为1小时~48小时,更优选为1小时~16小时。本发明提供一种依鲁替尼晶型1的又一制备方法(方法b),包括下列步骤:b1)将依鲁替尼粉末加入甲醇中,加热,搅拌使溶液澄清;b2)降温至室温,加入水,搅拌,析晶;b3)过滤收集固体,得到晶型1。其中:步骤b1)中,所述依鲁替尼采用与cn101610676a实施例1b相同的方法制备。步骤b1)中,所述依鲁替尼与甲醇的比例为50~250:1(mg/ml),优选为50~150:1(mg/ml),更优选为80~150:1(mg/ml);步骤b1)中,所述加热温度为40℃~65℃,优选为40℃~60℃,更优选为50℃;步骤b1)中,所述搅拌的时间为1~12小时,优选为1~5小时,更优选为1~3小时;步骤b2)中,所述室温为20℃~25℃,优选为25℃;步骤b2)中,所述降温在0.5小时~2小时内使温度降至所述室温,优选在1小时~2小时内将温度降至所述室温;步骤b2)中,所述水与步骤b1)中所述甲醇的体积比为1:1~1:10,优选体积比为1:1~1:6,更优选体积比为1:1~1:2;步骤b2)中,所述搅拌的时间为0.5小时~72小时,优选为1小时~48小时,更优选为1小时~16小时。本发明提供一种依鲁替尼新晶型的另一制备方法(方法c),包括下列步骤:c1)将依鲁替尼粉末加入甲醇和水的混合溶剂中,于适宜温度下搅拌,析晶;c2)过滤收集固体,得到晶型1。其中:步骤c1)中,所述依鲁替尼采用与cn101610676a实施例1b相同的方法制备。步骤c1)中,所述依鲁替尼与甲醇和水的混合溶剂总量的比例为50~250:1(mg/ml),优选为70~150:1(mg/ml),更优选为80~120:1(mg/ml);步骤c1)中,所述甲醇和水的体积比为1:1~10:1,优选体积比为1:1~6:1,更优选体积比为1:1~4:1;步骤c1)中,所述搅拌温度为0℃~45℃,优选为5℃~30℃,更优选为5℃~25℃;步骤c1)中,所述搅拌的时间为1小时~24小时,优选为3小时~16小时,更优选为5小时~10小时。本发明的有益效果为:本发明提供的依鲁替尼晶型1溶解性好,在生物介质中溶解度高,有利于提高药物的口服生物利用度,使药物获得更佳的治疗效果,有较好的临床应用价值。本发明提供的依鲁替尼晶型1具有良好的稳定性,并且几乎无吸湿性,确保药物在储存和运输过程中的稳定性以及临床应用中的安全性。本发明提供的依鲁替尼晶型1制备方法操作简单,成本低廉,对生产设备无特殊要求,宜于工业化生产,有较大的应用价值。附图说明图1为依鲁替尼晶型1的x射线粉末衍射(xrpd)图,其中横坐标为2θ值(度),纵坐标为强度(计数)。图2为依鲁替尼晶型1的差示扫描量热(dsc)分析图,其中横坐标为温度(℃),纵坐标为热流(瓦/克)。图3为依鲁替尼晶型1的热重分析(tga)图,其中横坐标为温度(℃),纵坐标为重量(%)。图4为依鲁替尼晶型1的动态水分吸附(dvs)分析图,其中横坐标为相对湿度rh(%),纵坐标为重量(%)。图5为25℃/40%rh条件下依鲁替尼晶型1的稳定性试验xrpd对比图,其中横坐标为2θ值(度),纵坐标为强度(计数),“0d”表示初始状态,“25℃-40%rh-5d-1”表示在25℃/40%rh条件下放置5天后的第1份样品,“25℃-40%rh-5d-2”表示在25℃/40%rh条件下放置5天后的第2份样品,其余标记的含义以此类推。图6为40℃/75%rh条件下依鲁替尼晶型1的稳定性试验xrpd对比图,其中横坐标为2θ值(度),纵坐标为强度(计数),“0d”表示初始状态,“40℃-75%rh-5d-1”表示在40℃/75%rh条件下放置5天后的第1份样品,“40℃-75%rh-5d-2”表示在40℃/75%rh条件下放置5天后的第2份样品,其余标记的含义以此类推。图7为25℃/40%rh条件下依鲁替尼晶型1的稳定性试验dsc对比图,其中横坐标为温度(℃),纵坐标为热流(瓦/克),各标记含义同图5。图8为40℃/75%rh条件下依鲁替尼晶型1的稳定性试验dsc对比图,其中横坐标为温度(℃),纵坐标为热流(瓦/克),各标记含义同图6。具体实施方式术语解释xrpd:x射线粉末衍射dsc:差示扫描量热分析tga:热重分析dvs:动态水分吸附分析hplc:高效液相色谱本发明所使用的仪器设备及方法参数:1.xrpd本发明所述的x射线粉末衍射(xrpd)图在brukerd8advancediffractometerx-射线粉末衍射仪上采集,测量方法参数为:x射线反射参数:cuk电压:40千伏特(kv)电流:40毫安培(ma)检测角度:3-40°2θ/3-30°2θ(热台xrd)扫描速度:0.2秒/步步长:0.02°2θ无反射样品板:24.6mmdiameterx1.0mmthickness(购自mticorporation)变温热台样品板:铜板(购自上海微图仪器科技发展有限公司)2.dsc本发明所述的差示扫描量热分析(dsc)图在tainstrumentsq200dsc差热分析扫描仪上采集,测量方法参数为:温度范围:25~300℃扫描速率:10℃/min保护气体:氮气气体流速:40ml/min3.tga本发明所述的热重分析(tga)图在tainstrumentsq500tga热重分析仪上采集,测量方法参数为:温度范围:室温~300℃扫描速率:10℃/min保护气体:氮气气体流速:40ml/min4.dvs本发明所述的动态水分吸附(dvs)分析图在tainstrumentsq5000tga动态水分吸附仪上采集,测量参数方法为:保护气体:氮气气体流速:10ml/min温度:25℃湿度范围:0%~80%步进速率:10%/120min5.hplc本发明所使用的高效液相色谱仪为ultimate3000,测量方法参数为:色谱柱:agilentc18(4.6*250mm*5μm)柱温:35℃流速:1.0ml/min检测波长:254nm进样量:5μl运行时间:60min配样溶剂:水/甲醇(20/80,v/v)进样浓度:1.0mg/ml流动相及运行梯度:流动相a:0.1%三氟乙酸水溶液;流动相b:0.1%三氟乙酸乙腈溶液实施例1:用作本发明新晶型(晶型1)制备的原料依鲁替尼粉末的制备采用与cn101610676a实施例1b相同的方法制备:向200ml三口瓶中加入3.86g的中间体(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺和35ml的二氯甲烷,开启搅拌。加入4.2ml三乙胺,缓慢滴加1ml的丙烯酰氯,加毕继续反应2小时。反应结束后,有机相分别用5%柠檬酸和食盐水溶液洗涤分层。有机层用mgso4干燥后旋干,残留物通过柱层析纯化(二氯甲烷/甲醇=25/1)得到2.15g依鲁替尼粉末。实施例2:依鲁替尼(1-((r)-3-(4-氨基-3-(4-苯氧基苯基)-1h-吡唑并[3,4-d]嘧啶-1-基)哌啶-1-基)丙-2-烯-1-酮)新晶型(晶型1)的制备将实施例1制备的依鲁替尼粉末500mg加入4ml甲醇中,加热至60℃搅拌1小时,得到澄清溶液。将该溶液冷却至25℃,滴加2ml水,在25℃下搅拌1小时后,在1小时内降温到5℃并继续搅拌48小时,过滤,收集固体并于50℃真空干燥18小时,得到466mg晶型1,其xrpd图谱如图1所示。实施例3:依鲁替尼晶型1的制备将实施例1制备的依鲁替尼粉末500mg加入4ml甲醇中,加热至50℃搅拌2小时,得到澄清溶液。将该溶液冷却至25℃,滴加2ml水,在25℃下搅拌2小时后,在1小时内降温到5℃并继续搅拌16小时,过滤,收集固体并于50℃真空干燥18小时,得到458mg晶型1,其xrpd图谱与实施例2所制备的产物的xrpd图谱相同。实施例4:依鲁替尼晶型1的制备将实施例1制备的依鲁替尼粉末500mg加入6ml甲醇中,加热至40℃搅拌2小时,得到澄清溶液。将该溶液冷却至25℃,滴加2ml水,在25℃下搅拌1小时后,在2小时内降温到5℃并继续搅拌1小时,过滤,收集固体并于50℃真空干燥18小时,得到435mg晶型1,其xrpd图谱与实施例2所制备的产物的xrpd图谱相同。实施例5:依鲁替尼晶型1的制备将实施例1制备的依鲁替尼粉末500mg加入4ml甲醇中,加热至50℃搅拌1小时,得到澄清溶液。将该溶液冷却至25℃,滴加4ml水,在25℃下搅拌3小时后,在1小时内降温到10℃并继续搅拌12小时,过滤,收集固体并于50℃真空干燥18小时,得到475mg晶型1,其xrpd图谱与实施例2所制备的产物的xrpd图谱相同。实施例6:依鲁替尼晶型1的制备将实施例1制备的依鲁替尼粉末500mg加入4ml甲醇中,加热至50℃搅拌3小时,得到澄清溶液。将该溶液冷却至25℃,滴加1ml水,在25℃下搅拌1小时后,在2小时内降温到5℃并继续搅拌16小时,过滤,收集固体并于50℃真空干燥18小时,得到463mg晶型1,其xrpd图谱与实施例2所制备的产物的xrpd图谱相同。实施例7:依鲁替尼晶型1的制备将实施例1制备的依鲁替尼粉末500mg加入4ml甲醇中,加热至60℃搅拌1小时,得到澄清溶液。将该溶液冷却至25℃,滴加2ml水,在25℃下继续搅拌48小时后,过滤,收集固体并于50℃真空干燥18小时,得到436mg晶型1,其xrpd图谱与实施例2所制备的产物的xrpd图谱相同。实施例8:依鲁替尼晶型1的制备将实施例1制备的依鲁替尼粉末500mg加入4ml甲醇中,加热至50℃搅拌2小时,得到澄清溶液。将该溶液冷却至25℃,滴加4ml水,在25℃下继续搅拌16小时后,过滤,收集固体并于50℃真空干燥18小时,得到442mg晶型1,其xrpd图谱与实施例2所制备的产物的xrpd图谱相同。实施例9:依鲁替尼晶型1的制备将实施例1制备的依鲁替尼粉末500mg加入4ml甲醇中,加热至50℃搅拌1小时,得到澄清溶液。将该溶液冷却至25℃,滴加2ml水,在25℃下继续搅拌1小时后,过滤,收集固体并于50℃真空干燥18小时,得到423mg晶型1,其xrpd图谱与实施例2所制备的产物的xrpd图谱相同。实施例10:依鲁替尼晶型1的制备将实施例1制备的依鲁替尼粉末500mg加入6ml体积比为2/1的甲醇/水混合溶剂中,在25℃下搅拌10小时,过滤,收集固体并于50℃真空干燥18小时,得到445mg晶型1,其xrpd图谱与实施例2所制备的产物的xrpd图谱相同。实施例11:依鲁替尼晶型1的制备将实施例1制备的依鲁替尼粉末500mg加入6ml体积比为2/1的甲醇/水混合溶剂中,在5℃下搅拌16小时,过滤,收集固体并于50℃真空干燥18小时,得到452mg晶型1,其xrpd图谱与实施例2所制备的产物的xrpd图谱相同。实施例12:依鲁替尼晶型1的制备将实施例1制备的依鲁替尼粉末500mg加入5ml体积比4/1的甲醇/水混合溶剂中,在25℃下搅拌5小时,过滤,收集固体并于50℃真空干燥18小时,得到440mg晶型1,其xrpd图谱与实施例2所制备的产物的xrpd图谱相同。实施例13:依鲁替尼晶型1的制备将实施例1制备的依鲁替尼粉末500mg加入6ml体积比为1/1的甲醇/水混合溶剂中,在10℃下搅拌10小时,过滤,收集固体并于50℃真空干燥18小时,得到464mg晶型1,其xrpd图谱与实施例2所制备的产物的xrpd图谱相同。实施例14:本发明依鲁替尼晶型1的溶解度试验1)ph1.8的sgf(人工胃液)配制取浓盐酸0.765ml,加水约80ml、胃蛋白酶1.0g、氯化钠0.2g,摇匀后,加水稀释成100ml,用盐酸水溶液调ph至1.8。2)ph5.0的fessif(进食状态下的人工肠液)配制取0.865g冰乙酸、0.831g牛胆酸钠、0.288g卵磷脂、1.520g氯化钾置于100ml容量瓶中,用水溶解并定容至100ml,用氢氧化钠水溶液调ph至5.0。3)对照品依鲁替尼晶型a的制备向50ml反应瓶中加入400mg按照实施例1所述方法制备的依鲁替尼粉末、5.0ml的80%丙酮水溶液、10mg的活性炭,加热到45℃,搅拌30分钟,过滤除去活性炭,并用80%丙酮水溶液淋洗滤饼。将滤液温度调整到30±3℃,在2小时内滴加水2.5ml,再搅拌1小时,缓慢冷却至5±2℃,搅拌2小时后有固体析出,过滤,滤饼用60%丙酮水溶液洗涤淋洗,收集固体并于45℃真空干燥20小时,得到364mg依鲁替尼结晶,其xrpd图谱与cn104736178a的附图1相同,因而该晶型确证为依鲁替尼晶型a。4)测定方法与结果取按照本发明实施例2制备的依鲁替尼晶型1样品,以及依鲁替尼晶型a对照品各10mg,分别在25℃的水3ml、sgf溶液3ml(ph1.8)及fessif溶液3ml(ph5.0)中各自搅拌24小时,形成饱和溶液,过滤后通过hplc进行溶解度检测。以依鲁替尼晶型a的已知浓度的溶液为标准外标物,获得峰面积值,通过峰面积比值得到本发明晶型1和cn104736178a中的晶型a分别在25℃的水、sgf溶液(ph1.8)及fessif溶液(ph5.0)中的溶解度(表1)。结果显示,本发明晶型1在水、sgf溶液(ph1.8)及fessif溶液(ph5.0)中的溶解度分别为1.24μg/ml、562.34μg/ml和229.50μg/ml,而cn104736178a中的晶型a在水、sgf溶液(ph1.8)及fessif溶液(ph5.0)中的溶解度仅分别为1.01μg/ml、375.83μg/ml和125.45μg/ml。可见,本发明晶型1的溶解度优于cn104736178a中的晶型a。表1本发明晶型1与cn104736178a中的晶型a的溶解度对比实施例15:本发明依鲁替尼晶型1的引湿性试验取按照实施例2制备的本发明晶型1的样品10mg,采用动态水分吸附(dvs)仪在25℃下于0%rh~80%rh相对湿度范围内测试该晶型的引湿性,并采用表2所示的标准(中国药典2015版第四部通则9103-引湿性试验指导原则)判断引湿特征。表2引湿性判断标准引湿特征增重界定不吸湿不高于0.2%轻微吸湿高于0.2%,但不高于2.0%易吸湿高于2%,但不高于15%极易吸湿高于15%本试验的dvs图如图4所示。结果表明,本发明晶型1在0%rh至80%rh范围内的重量变化为0.5%,几乎不吸湿。实施例16:本发明依鲁替尼晶型1的稳定性试验1)测定方法取按照实施例2和实施例10制备的本发明晶型1的两份样品各20mg分别置于表面皿内,在25℃/40%rh、40℃/75%rh这两个条件下,于第5天、第10天、第30天取样。考察样品外观变化,利用xrpd观察晶型、dsc观察熔点等表征数据变化,并通过hplc检测考察主峰及杂质变化情况。2)测定结果外观:两份样品在25℃/40%rh、40℃/75%rh两个条件下30天均无变化,为白色粉末。xrpd:两份样品在25℃/40%rh、40℃/75%rh两个条件下30天未检测到xrpd图谱(结晶度和晶型)有明显变化,具体结果见图5和图6。dsc:两份样品在25℃/40%rh、40℃/75%rh两个条件下30天未观察到dsc图谱(熔点)有明显变化,具体结果见图7和图8。hplc:两份样品在25℃/40%rh、40℃/75%rh两个条件下30天基本没有分解,仍然保持很高的化学纯度,具体结果见表3。表3本发明晶型1两份样品的稳定性试验hplc检测结果结果表明,本发明晶型1的两份样品在25℃/40%rh、40℃/75%rh两个条件下30天在晶型、熔点和化学稳定性等方面均无明显变化,稳定性良好。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1