基因倒位突变检测方法与流程
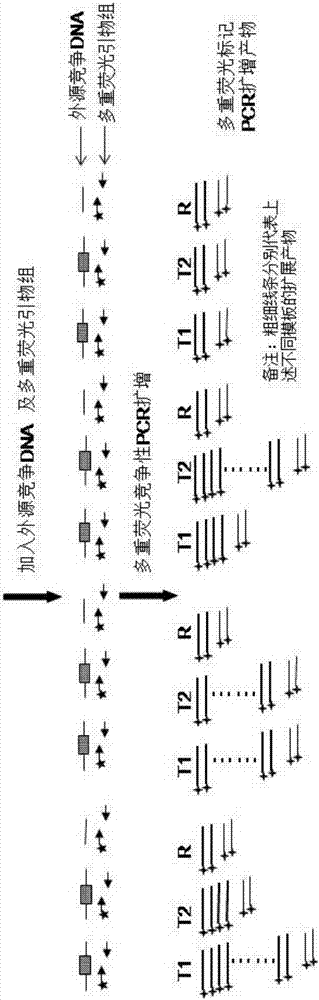
本发明涉及基因检测领域的一种基因倒位突变检测方法。
背景技术:
染色体的断裂及断裂后染色体断端的异常重接是引起染色体结构畸变的遗传学基础,而倒位(inversion)是常见的一种染色体结构畸变类型。倒位指染色体上某一区段连发生两次断裂,中间的片段发生180度的倒转,然后重接。很多染色体倒位并没有改变基因的数目,也没有对断点附近的基因造成影响,因此,该类突变的携带者拥有正常的表型,但较大区域的倒位突变可能会在减数分裂中影响同源染色体的配对从而造成杂合子个体生育能力下降。但有些倒位突变的断点位于功能基因内部或调控区附近,会影响改基因的正常表达从而引起疾病(如f8基因22号及1号内含子倒位突变)。
对染色体倒位的快速而准确的检测将有助于医学和遗传学相关领域的科学研究和疾病诊断。现有的具体检测方法可以分为两大类,一类是全基因组水平上的染色体倒位突变检测,另一类是对目标区域进行倒位突变检测。前者主要为细胞核型分析,该技术是检测染色体病最常见的方法,即将待测的细胞的染色体按照该其固有的染色体形态特征和规定,进行配对、编号和分组,并进行形态分析的过程,该技术在染色体疾病诊断中占据重要地位。但其存在分辨率较低,操作复杂,需要专业人员判读检测结果,需要活细胞培养等不足之处,限制了该技术在临床染色体倒位检测中的应用。而后者主要针对特定目标区段的检测,包括southern印迹杂交(southernblot)技术(lakichdetal.,1993)、荧光原位杂交技术(fluorescentinsituhybridization,fish)(sheencretal.,2011)、倒位突变等位基因特异pcr(liuqeta.l,1997)以及反向pcr技术(inverse-pcr)(rossettietal.,2005)。
southern印迹法的基本原理是:具有一定同源性的两条核酸单链在一定的条件下,可按碱基互补的原则特异性地杂交形成双链。一般利用琼脂糖凝胶电泳分离经限制性内切酶消化的dna片段,将胶上的dna变性并在原位将单链dna片段转移至尼龙膜或其他固相支持物上,经干烤或者紫外线照射固定,再与相对应结构的标记探针进行杂交,用放射自显影或酶反应显色,从而检测特定dna分子的含量。由于染色体倒位会导致基因序列发生变化,使用特定的限制性内切酶如bclⅰ等对发生倒位的基因组进行酶切时,产生的酶切片段大小会与正常基因组酶切后片段大小不一致,并可通过southern印迹法检测出来。southern印迹法的应用非常广泛,并被最早使用于血友病f8基因的倒位检测中,是该疾病检测倒位的金标准,具有高度特异性及灵敏性。其缺点在于操作复杂,耗时约一周。之前采用放射性标记物,其灵敏度极高,但是存在着放射性污染及放射核素发生衰变所产生的不稳定性两大缺点。目前多采用地高辛代替放射性标记物,以避免污染。
荧光原位杂交技术(fish,fluorescenceinsituhybridization)是一种重要的非放射性原位杂交技术,也可用于染色体的倒位检测。它的基本原理是:如果被检测的染色体或dna纤维切片上的靶dna与所用的核酸探针是同源互补的,二者经变性-退火-复性,即可形成靶dna与核酸探针的杂交体。将核酸探针的某一种核苷酸标记上报告分子如生物素、地高辛,可利用该报告分子与荧光素标记的特异亲和素之间的免疫化学反应,经荧光检测体系在镜下对待测dna进行定性、定量或相对定位分析。以f8基因第22号内含子倒位为例,根据同源重组区域位置需要设计三种bac克隆探针,rp11-54i20(insertsize179.3kb),rp11-296n8(178.7kb),rp11-405n23(164kb),其中rp11-54i20探针可与倒位区域上游1.25mb区域杂交,而rp11-296n8与rp11-405n23则可与倒位发生区域杂交,且为区别信号这两个探针杂交位置相差150kb,三种探针标记为不同的荧光信号,分别为蓝(b)、红(r)、绿(g),杂交后通过检测荧光信号就能知道是否有倒位发生。若为野生型则荧光信号为brg,若为倒位则为bgr,携带者则这两种组合的信号都会出现bgr/brg。荧光原位杂交技术检测的结果判读直观快速,具有较高的敏感性与特异性,但也存在一些不足。fish探针的制备比较耗时与昂贵;对实验操作的技术性要求比较高;由于染色体形态问题,有可能出现无法判读的情况;且该方法对于男性样本的阳性检出灵敏度要高于女性样本;一般从样本制备到检测出结果需要三天时间,耗时较长。
倒位突变等位基因特异pcr是指针对已知倒位突变断点两侧序列设计一组pcr引物可以对野生型和突变型等位基因同时进行扩增,但扩增产物在长度上有一定差异,通过常规琼脂糖凝胶电泳即可分离。在某些情况下,为了区分野生型和突变型等位基因,扩增产物片段会很大(如大于5kb),这时pcr扩增体系需要采用ld-pcr(长距离pcr)。ld-pcr是一种能扩增5kb以上基因片段的方法。与常规pcr相比,其对扩增区域变化的适应性强,在临床中尤其适合于短核苷酸动态突变疾病和基因序列重复异常扩展相关疾病的基因诊断,也被用于染色体或基因倒位突变的检测中。以血友病f8基因倒位突变检测为例,根据该疾病相关的基因组同源重组区域的不同有两种类型,在重组区域两端设计多组引物,在同一体系进行ld-pcr反应,最终通过琼脂糖凝胶电泳检测扩增所得产物的片段大小,即可判断是否有倒位发生以及是哪种类型的倒位。ld-pcr是目前应用最多的对染色体特定区域的倒位检测方法,其优势在于检测步骤简单,只需在一个体系中进行ld-pcr反应,最后跑电泳检测条带大小即可。对于ld-pcr扩增技术,其缺点是难度较普通pcr大,且需要高质量的dna提取,才能提供扩增所需的完整模板。由于ld-pcr扩增后的产物比较大,电泳检测需要的时间更长,如对血友病f8基因倒位的ld-pcr检测,仅对pcr产物进行电泳的耗时一般就需要12小时。
倒位pcr技术(inversion-pcr)是为了解决ld-pcr扩增片段长,基因扩增难度大的缺陷而开发出来的。该技术是应用特定的限制行内切酶对基因组进行酶切,酶切片段包含需要检测的染色体倒位区域的同源重组序列,酶切后的dna片段用t4连接酶自连接成环状,然后在连接点两端设计引物进行pcr扩增,最终经琼脂糖凝胶电泳检测,根据扩增产物大小即可判断是否有倒位及倒位类型。以血友病f8基因倒位检测为例,倒位pcr技术先应用bclⅰ限制酶切f8基因,酶切片段包含同源重组序列,酶切后的dna片段用t4连接酶自连接成环状结构,然后在连接点两端设计四个引物进行pcr扩增,根据是否为倒位以及倒位类型不同,4个引物的扩增产物及其长度也不同,最终扩增产物经琼脂糖凝胶电泳检测即可判断是否有倒位及倒位类型。该方法需要通过酶切、连接和pcr扩增三步操作,与ld-pcr相比大大减小了扩增难度,但步骤较ld-pcr复杂,所需dna用量大,需要的检测时间也要更长一些。
多重荧光竞争性pcr拷贝数定量技术是上海天昊生物科技有限公司于2010年10月申请的一项发明专利(专利号:zl201010180551.2),该技术是基于竞争性pcr原理并结合多重荧光pcr扩增以及毛细管电泳长度差异片段分离技术对参照基因和多个目标基因同时进行定量,并利用参照基因对检测结果进行校正后获取多个目标基因的正确拷贝数,具体流程如下:针对多个目标基因以及参照基因进行多重pcr引物设计,每对引物中一条将标上荧光引物,同种荧光引物对应扩增产物长度必须有差别至少5bp以上;针对每个基因的扩增片段,合成或克隆获得对应片段引物间序列缺失或插入少数几个碱基(通常为2)的dna片段作为竞争dna,并进行精确定量;将适量样本dna与适量竞争dna混匀,混入的每种竞争dna的分子数量尽可能一致并估计与样本dna中2拷贝基因的分子数量大约一致;对样本/竞争dna混合物进行多重荧光pcr扩增;扩增产物通过变性毛细管电泳将不同长度的片段分开,收集每个条带的位置信息以及荧光强度(这一步通常在一代测序仪上完成如美国应用生物公司的测序仪abi3130,abi3500以及abi3730等);由于不同基因的扩增产物在长度或荧光标记上不同,而且同一基因位点上样本dna以及竞争dna扩增产物长度也不同,因此根据毛细管电泳峰形图可以明确每个基因位点上样本dna以及竞争dna各自扩增产物量,计算每个基因位点的样本dna峰(s峰)与竞争dna峰(c峰)的峰高或面积比(s/c);将每个目标基因位点的s/c比值分别除以参照基因的s/c比值获得该目标基因位点的r值;将该r值除以正常参照样本的r值(或所有样本的r值中位数)再乘以正常参照样本(或绝大多数样本预计)在该位点的拷贝数即可获得检测样本在该基因位点上的拷贝数;如果一个检测体系有多个参照基因片段,每个目标基因片段可针对每个参照基因片段数据分别计算获得多个检测值,然后取其平均数或中位数作为检测样本在该基因片段上的拷贝数。
技术实现要素:
本发明为了实现对基因倒位突变进行快速而准确地检测,提供一种快速简单的染色体倒位突变检测方法,该方法基于倒位突变等位基因特异性pcr预扩增和多重荧光竞争性pcr拷贝数定量技术原理开发而成。首先利用常规ld-pcr体系在较低pcr循环数下获得预扩增产物,然后采用多重荧光竞争性pcr拷贝数定量技术对预扩增产物进行野生型特异扩增片段以及突变型特异扩增片段同时进行拷贝数定量,根据预扩增片段的扩增谱来确定样本的基因型。
为实现上述技术目的,达到上述技术效果,本发明通过以下技术方案实现:
一种基因倒位突变检测方法,包括以下步骤:
1)针对待测的基因组倒位突变区域,在区域内外设计对应的多组pcr引物,当检测样本为正常样本和发生不同类型基因组倒位突变的样本时,所述pcr引物的扩增产物在碱基序列上会存在差异;
2)根据需要检测的基因组倒位突变情况,将所述多组pcr引物在一个或多个反应体系内对待测样本进行低循环pcr预扩增获得预扩增产物;
3)针对所述pcr扩增产物,区分为倒位突变的野生型等位基因和突变型等位基因的差异碱基序列,设计对应的多重pcr引物,每对引物中的一条具有荧光标记;在倒位突变区以外的其他基因组区域选取至少一个参照基因片段,设计至少一对pcr引物,每对引物中的一条具有荧光标记,同种荧光标记的多重pcr引物对应的扩增产物的长度有差异;
4)设计针对上述标记dna序列和参照基因序列的外源竞争dna片段,所述外源竞争dna片段与步骤3中对应的多重荧光pcr引物的扩增产物高度一致,为对应扩增产物的序列缺失或插入少数1-50个碱基的序列,该外源竞争dna片段可以被步骤3中对应pcr引物扩增;
5)对所有外源竞争dna片段进行定量;
6)将混合后的外源竞争dna片段与一定量的步骤2中pcr预扩增产物或一定量的未扩增的正常样本基因组dna混合,以二者的混合dna片段为模板进行多重荧光pcr扩增;
7)扩增产物通过变性荧光毛细管电泳将不同长度的片段分开,得到每个条带的位置信息以及荧光强度;
8)根据数据分析预扩增片段的扩增谱来确定样本的基因型。
进一步的,所述步骤2中低循环pcr预扩增的pcr循环数在40个循环以内。
优选的,所述步骤2中低循环pcr预扩增的pcr循环数为10-20个循环。
优选的,所述步骤3中的同种荧光标记的多重pcr引物对应的扩增产物的长度差异大于5bp。
优选的,所述步骤4中对应扩增产物的序列缺失或插入2-5个碱基的序列。
优选的,所述步骤5中对所有外源竞争dna片段进行定量后按一定比例混合。
进一步的,所述步骤8中数据分析包括:
a)计算每个dna序列的样本条带(s)/外源竞争dna(c)条带的荧光峰高或面积比(s/c);
b)将标记dna序列的s/c比值除以参照基因dna序列的s/c比值获得该标记dna序列的r值;
c)该r值除以未扩增的正常样本基因组dna的r值再乘以未扩增的正常样本基因组dna在该标记dna序列上的拷贝数即可获得检测样本pcr预扩增产物在该标记dna序列上的拷贝数,该拷贝数的大小可指示标记dna序列在步骤2中是否获得了显著性pcr扩增,显著性pcr扩增是指计算标记dna序列的拷贝数显著性高于线性扩增理论拷贝数;
d)若标记dna序列的拷贝数高于线性扩增理论拷贝数的2倍即定义为标记dna序列获得显著性pcr扩增;
e)基因型判定:若检测样本的野生型相关标记dna序列获得显著性pcr扩增的,判定该样本的倒位突变基因型为野生型;
若检测样本的突变型相关标记dna序列获得显著性pcr扩增的,判定该样本的倒位突变基因型为突变型;
若检测样本的野生型和突变型相关标记dna序列均获得显著性pcr扩增的,判定该样本的倒位突变基因型为杂合型。
本发明的有益效果是:
相对现有其他检测方法,等位基因特异性pcr扩增后进行琼脂糖电泳分离判定是比较简单快速的方法,但当扩增产物片段过大时,这种方法会由于较长时间的pcr扩增过程,长时间的电泳分离时间,野生型和突变型特异扩增产物片段不易区分以及这两种长度不等片段不均衡扩增导致判定错误等缺点。本发明通过不要求野生型和突变型特异扩增产物片段大小有区别,减少pcr扩增循环数,去除琼脂糖电泳以及对扩增产物量进行准确定量等操作,有效克服了传统等位基因特异性pcr检测方法的缺点,具备了检测更快速、操作更简单以及检测更准确的优点。此外,由于低循环预扩增以及后续荧光竞争性pcr均可以进行多重pcr扩增,而且不要求野生型和突变型特异扩增产物片段大小有区别,所以该发明可以对多个倒位突变同时进行检测。
上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合附图详细说明如后。本发明的具体实施方式由以下实施例及其附图详细给出。
附图说明
此处所说明的附图用来提供对本发明的进一步理解,构成本申请的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
图1a为基因组倒位突变检测原理图一;
图1b为基因组倒位突变检测原理图二;
图1c为基因组倒位突变检测原理图三;
图2为血友病f8基因内含子22倒位突变检测原理。
具体实施方式
下面将参考附图并结合实施例,来详细说明本发明。
图1a、图1b、图1c依次为本发明基因组倒位突变检测原理图,以下以f8基因内含子22倒位突变的检测应用作为该发明的实施案例。
一、背景介绍
血友病a是最常见的x连锁隐性遗传性出血病。目前已报道的导致血友病a的f8基因(f8)突变有1200多种,包括错义突变,无义突变,剪接位点突变,大缺失,小缺失或插入突变及dna重组等。约2/3的患者有明确的家族史。其中内含子22倒位(inv22)是最重要的热点突变,约占重型血友病a的50%左右。f8基因位于x染色体xq28,其22号内含子在所有内含子中最长,约32.8kb,其中9.5kb的序列片段(int22-1)与距离f8基因上游约400kb和500kb的两个片段(int22-2和int22-3)高度同源性。内含子22倒位主要分为远端即typei倒位,近端即typeii倒位两种类型,还有少见类型即缺失型22倒位(具有致病性)及重复型22倒位(可能不具有致病性)等。int-3与int22-1反向,其同源重组导致了typei22倒位(占大多数),而int22-2和int22-3两个同源序列互换后导致int22-2与int22-1反向,其重组后导致了typeii22倒位(约为typeiinv22的1/5)。如果int22-1与其同向的int22-2或int22-3同源序列发生重组则可能产生缺失或重复,而缺失可以发生在x染色体内或染色体间,而重复只能发生在染色体间。
二、检测体系设计
2.1检测体系设计方案
具体实验设计方案如下:
1)根据f8基因倒位突变情况和类型,针对3个同源片段(如图2中的int22-1,int22-2和int22-3),在区域内外设计对应的5条pcr引物(如图2中f8a1f,f8a1r,f8a2r,f8a2fa3r,f8a3f),不同倒位情况和类型下进行ld-pcr得到的扩增产物的dna序列必须存在差异。
2)根据f8基因需要检测的基因组倒位突变情况,将所述5条ld-pcr引物在两个个反应体系内对待测样本进行低循环pcr预扩增获得预扩增产物,其中一个反应体系使用f8a1f,f8a1r,f8a2r,f8a3f共4条ld-pcr预扩增引物,另一个反应体系使用f8a2fa3r,f8a3f,f8a1r共3条ld-pcr预扩增引物,pcr循环数控制在12-16个循环之间;
3)针对所述pcr扩增产物中可以区分倒位突变的野生型等位基因和突变型等位基因的差异碱基序列(以下简称标记dna序列,图2中使用m1,m2,m3标记),设计对应的三对多重pcr引物,每对引物中的一条具有fam荧光标记;在倒位突变区以外的其他基因组区域选取四个参照基因片段,各设计一对pcr引物,每对引物中的一条具有fam荧光标记;上述同种荧光标记的多重pcr引物对应的扩增产物的长度至少有5bp差异,具体扩增长度见表4;
4)设计针对上述标记dna序列和参照基因序列的外源竞争dna片段,该外源竞争dna片段与(3)中对应的多重荧光pcr引物的扩增产物高度一致,为对应扩增产物的序列缺失2个碱基的序列,该外源竞争dna片段可以被(3)中对应pcr引物扩增;
5)对所有外源竞争dna片段进行定量后按一定比例混合;
6)将混合后的外源竞争dna片段与一定量的(2)中pcr预扩增产物及或一定量的未扩增的正常样本基因组dna混合,以二者的混合dna片段为模板进行多重荧光pcr扩增,pcr体系中还包含一对性别位点(amelo)扩增引物;
7)扩增产物通过变性荧光毛细管电泳将不同长度的片段分开,得到每个条带的位置信息以及荧光强度;
8)数据分析:计算每个dna序列的样本条带(s)/外源竞争dna(c)条带的荧光峰高或面积比(s/c),然后将标记dna序列的s/c比值除以参照基因dna序列的s/c比值获得该标记dna序列的r值;该r值除以未扩增的正常样本基因组dna的r值再乘以未扩增的正常样本基因组dna在该标记dna序列上的拷贝数即可获得检测样本pcr预扩增产物在该标记dna序列上的拷贝数,该拷贝数的大小可指示标记dna序列在(2)中是否获得了显著性pcr扩增,显著性pcr扩增是指计算标记dna序列的拷贝数显著性高于线性扩增理论拷贝数,通常如果标记dna序列的拷贝数高于线性扩增理论拷贝数的2倍即定义为标记dna序列获得显著性pcr扩增;
9)基因型判定:根据三个标记dna序列m1,m2,m3是否获得显著性pcr扩增可对检测样本进行倒位突变基因型的判定,判定标准如表1所示。
10)性别判定:根据性别位点扩增产物峰进行性别判定,如果存在2个峰为男性,如果存在1个短片段峰,为女性。
图2所示为本实施例中对血友病f8基因内含子22倒位进行ld-pcr的原理图。a,b分别为正常人中的两种基因型,c为血友病内含子22倒位ⅰ型,d为血友病内含子22倒位ⅱ型。
表1基因型判定标准
2.2检测体系原材料的选择及引物、荧光引物和外源竞争型dna片段的设计
2.2.1原材料的选择
本检测体系中所涉及到的主要原材料的生产厂家如表2所示。
表2检测体系使用的主要原材料
2.2.2引物、荧光引物和外源竞争型dna片段的设计
根据染色体倒位分型原理(见图2)设计5条ld-pcr引物(f8a1f,f8a1r,f8a2r,f8a2fa3r,f8a3f),分为两组不同的ld-pcr引物混合液,其中ld-pcr引物混合液a1包括:f8a1f,f8a1r,f8a2r,f8a3f共4条ld-pcr预扩增引物,每条引物浓度各2μm。ld-pcr引物混合液a3包括:f8a2fa3r,f8a3f,f8a1r共3条ld-pcr预扩增引物,每条引物浓度各2μm。引物序列信息如表3。
表3ld-pcr引物序列表
根据染色体倒位分型原理(见图2)针对三个目标dna序列以及四个参照dna序列进行多重pcr引物设计,另外加入了amelo基因的扩增引物用于性别鉴定,以进一步提高检测的准确性。所述的每对引物中有一条为5’端fam荧光素标记。以上所述引物对混合组成pcr引物混合液,每条引物浓度各2μm。所述多重荧光pcr引物序列及相关补充信息请见表4和表5。
表4多重荧光pcr引物序列表
表5多重荧光pcr引物补充信息表
*homosapiensgrch37.p10(accessiongcf_000001405.22)
针对每个dna序列的扩增片段,设计与对应引物扩增产物序列对比缺失或插入2个碱基的dna片段作为外源竞争dna,并进行精确定量,适量外源竞争dna混匀,混入的每种外源竞争dna的分子数量相等并估计与ld-pcr扩增产物dna中参照dna序列的分子数量一致,得到外源竞争dna混合液。外源竞争dna由苏州金唯智进行克隆合成。
多重荧光竞争性pcr目标位点序列,参照基因及外源竞争dna序列信息:
m1(标记位点)
m1外源竞争dna序列,94bp
tcaaggcaacacaaatgagaatagcaataatcatataacgattggaaaatatagcctagtatctctctaaataaagcacatggctcatatggct
m2(目标位点)
m2外源竞争dna序列,80bp
gtgcccacttttataccagttccatactgttttagtaactatagccttattataaattgaagtccagtaatgtcatgcct
m3(目标位点)
m3外源竞争dna序列,142bp
acaaccagagcagaaatcaatgaaatacaaaagattaaaaacacaagaaattttaaaagtcaatagcgtcattgaaatgaatactctgacaagtcagcaacaaaatgaatcaaaaaaaacaaaaaggagtgaggaaacacaa
参照基因a
参照基因a外源竞争dna序列,73bp
tgagccaaaaattcagaatacaaggagctttaggaaaaaggctgtatgctgcgtttgcctgccttccaagcaa
参照基因b
参照基因b外源竞争dna序列,143bp
cactgagccccagagacctgacaagcctttgagccgtgcctgaaaaatgtggctcatcctcaggctgcagcggggaaaatggaagagtttaattggttctgacttcaggattcagatcatagttttccctggaggtgtgcatt
参照基因c
参照基因c外源竞争dna序列82bp
cccaccagagcttcccttagcaccaggtggcagctggttcattttgccaccctccagtagccttttcctggtggttctgcat
参照基因d
参照基因d外源竞争dna序列,178bp
ccaccaagctgatgtgttccttagcactgatggcaattctcttcagtctggggacaggtatcatctcctttggtttatcagtggacggagcacgggctgcagggttcaacaacatccatgtcctcaaagaaggcgcagggttctagcatgtggccatttctcacctttcgatagcttt
三、样本检测实例
3.1样本信息及质控
选择2个基因型已知患者样本进行测试,样本a为男性甲型血友病患者;样本b为女性携带者。
分别将待测样本a和待测样本b及一个已知正常的男性参照样本dna用biophotomerterplus(eppendor)(核酸蛋白测定仪)进行精确定量并稀释至100ng/μl待用。
3.2ld-pcr扩增反应
每个样本需进行2个ld-pcr反应,每个反应各需1μl样本。
ld-pcr体系(20μl)配置:
ld-pcr反应条件设置如下:98℃1min;12个循环(98℃10秒,68℃15min+20s/循环),在72℃延伸10分钟,4℃保存;
3.3多重荧光pcr扩增反应
各取1μlld-pcr产物,加入9μlddh2o,98℃5min后用作多重荧光pcr的模板。
多重荧光pcr体系(20μl)配置:
pcr反应条件设置如下:95℃5min;7个循环的touchdown程序(95℃20秒,64℃-0.5℃/循环40秒和72℃1.5min),28个循环扩增(95℃20秒,60℃30秒和72℃1.5min),在72℃延伸2分钟,在60℃延伸60分钟,4℃保存。
3.4毛细管电泳
pcr反应完成后,将pcr扩增产物稀释5-20倍,取出1μl加入8.9μlhi-di(高度去离子甲酰胺)和0.1μlliz500(abi公司专利染料,被用来标记分子量内标),混匀后95℃5min,置于3130xl测序仪上进行毛细管电泳。
结果使用genemapper4.0软件进行数据分析,读取峰高等数据,参照附录图所示。
数据分析表
1)表6为2个检测样本及1个正常样本的3个位点毛细管电泳峰高数据。
表6毛细管电泳峰高数据结果
2)样本dna峰高值s除以外源竞争dna峰高值c
计算每个位点的样本dna峰(s峰)与外源竞争dna峰(c峰)的峰高比(s/c),得到结果如表7所示。
表7峰高比(s/c)数据结果
3)样本标记位点的拷贝数
将每个标标记位点的s/c比值除以参照位点的s/c比值获得该标记位点的r值;该r值除以未扩增的正常样本基因组dna的r值再乘以未扩增的正常样本基因组dna在该标记位点上的拷贝数即可获得检测样本pcr预扩增产物在该标记位点上的拷贝数,得到结果如下表所示。
表8样本检测结果
标记位点获得了显著性pcr扩增的判断标准为该位点拷贝数的大小是否高于线性扩增理论拷贝数的2倍,即在12个循环ld-pcr扩增反应条件下,标记位点拷贝数大于24即可判定为显著性pcr扩增。根据表1基因型判定标准,对样本检测结果进行基因型判定。
四、结论
样本a检测结果为f8基因第22内含子i型倒位的男性,与实际结果一致。
样本b检测结果为f8基因第22内含子i型杂合的女性,与实际结果一致。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!